


 红肩章7076557
红肩章7076557
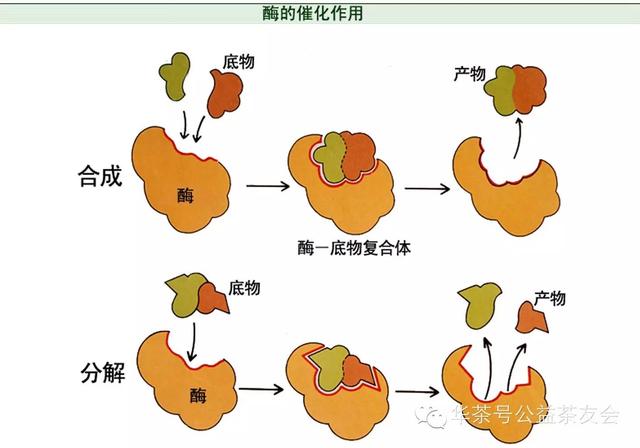
范文一:固体碱催化剂
?594?化学通报 2002年第9期 http:ΠΠhxtb.icas.ac.cn
固体碱催化剂
魏 彤 王谋华 魏 伟 孙予罕 钟 炳
(中国科学院山西煤炭化学研究所国家重点实验室 太原 030001)
摘 要 综述了最近30年来固体碱催化剂的研究现状,包括固体碱催化剂的种类、优缺点以及各
类催化剂中影响其催化性能的因素。重点探讨了碱位前驱体和载体对负载型无机固体碱碱强度的影响。展望了未来固体碱化剂的发展方向。
关键词 固体碱 碱性分子筛 碱催化
Abstract Thedevelopmentofsolidbasecatalystswas,mer2
itsanddrawbacksofsolidbaseaswellbehaviorofsolidbasewerepresentedindetail.ofsupportonthebasestrengthofsolidbaseweredis2cussed.Atofsolidbasecatalystinthefutrurewaspresented.
Keyw base,Basiczeolite,Basiccatalysis
随着世界环保意识的加强以及绿色化学的发展,人们越来越重视环境友好的催化新工艺过程。以固体碱作为催化剂具有高活性、高选择性、反应条件温和、产物易于分离、可循环使用等诸多优点,尤其在精细化学品合成方面可使反应工艺过程连续化,增强了设备的生产能力,发挥着越来越明显的优势,可望成为新一代环境友好的催化材料
化剂的研究起步较晚,发展也比较缓慢
在积极研制开发阶段。
自20世纪50年代固体碱催化剂引起科学家们的重视以来[5][3,4][1,2]。然而,相对固体酸催化剂而言,对固体碱催,主要原因在于固体碱,尤其是超强固体碱催化剂制备复杂、成本昂贵、强度较差、极易被大气中的CO2等杂质污染,而且比表面都比较小。因此,各国都处,已经发展了多种类型的固体碱催化体系,按照载体和活性位的性质不同,固体碱大体可分为有机固体碱,有机无机复合固体碱,以及无机固体碱,其中无机固体碱又可分为金属氧化物型和负载型。但总的来看,固体碱催化剂的研究尚缺乏系统性。
1 有机固体碱
通常有机固体碱主要是指端基为叔胺或叔膦基团的碱性树脂类固体碱,例如端基为三苯基膦的苯乙烯和对苯乙烯共聚物
子[7][6]。氮和磷的最外层分别有两个成对的s电子和三个未成对的p电33
3。在胺或膦分子中,氮或磷原子是sp杂化的,其中三个未成对电子分别占据三个sp轨道,每一个轨道和其它碳原子或氢原子的轨道成键,第四个sp轨道含有一对孤对电子,在棱锥形胺或膦
分子的顶点。就是这一对孤对电子使有机胺或有机膦呈不同强度的碱性。这种有机固体碱的优点是碱强度均一,但是它热稳定性不好,只能适用于低温反应,且制备复杂,成本较高。
魏 彤 女,26岁,博士生,现从事固体碱的制备及应用研究。
国家杰出青年基金资助项目(29625307)
2001212221收稿,2002203211修回
http:ΠΠhxtb.icas.ac.cn 化学通报 2002年第9期?595?2 有机无机复合固体碱
目前所研制的有机无机复合固体碱主要为负载有机胺或季铵碱的分子筛[8]。负载有机胺分子筛的碱活性位主要是能提供孤对电子的氮原子,而负载季铵碱分子筛的碱活性位主要是氢氧根离子。由于这类固体碱的活性位是以化学键和分子筛锚接的有机碱,所以反应过程中活性组分不会流失,而且碱强度均匀。同时也是由于其活性位为有机碱,故而不能适用于高温反应,且无法制备出强碱性的固体碱。
3 无机固体碱
无机固体碱催化剂因其制备简单、碱强度分布范围宽、向。无机固体碱主要包括金属氧化物、3.1 金属氧化物型
以氧化镁为例[9],低温处理时,其表面的碱性位主,其表面产生面、线、点等缺陷,使原来六配位的氧变成五配位、四配位或者三配位,增加了氧原子的电荷密度,从而使氧化镁表面带上不同程度的强碱性位。
一般而言,碱金属和碱土金属氧化物催化剂的碱性强度随碱金属和碱土金属的原子序数的增加而增加[10],其顺序为Cs2O>Rb2O>K2O>Na2O;BaO>SrO>CaO>MgO。另外煅烧温度和前驱体
[10]的种类也显著影响碱金属及碱土金属氧化物的碱强度,通常煅烧温度高有利于得到强的碱性位,而不同前驱体煅烧所得碱土金属氧化物的碱强度顺序为碳酸盐>氢氧化物>醋酸盐。总之,通过
改变制备条件或选择不同的前驱体,可以制备出具有强碱位甚至超强碱位的碱金属及碱土金属氧化物,但是这些固体碱均为粉状,没有机械强度,不易从产物中分离,而且其比表面积小(除MgO外均小于70mΠg)2[11]。此外,其比表面积随碱性位增强而显著降低。例如对于碱土金属氧化物而言,其比表面积的大小顺序为MgO>>CaO>SrO>BaO。这些都限制了它们在工业催化中的应用。
3.2 水合滑石类固体碱催化剂
水合滑石类材料是层柱双氢氧化物
M1=Mg、Zn或Ni,M2=Al、Cr或Fe,Am-[12],其结构式为[M1-2-
n-2+M23+-(OH)2(x+1)](A1mnH2O,其中Πm)?可以是Cl,CO32+3+等。通常以M和M为中心的M(OH)6八面体单元通过共边形成带有正电荷的层板,而A
分子,An-和H2O分别是位于层板间的各种阴离子和水起平衡层板正电荷的作用[12]。当M1为Mg、M2为Al时,这种水滑石类催化剂表面同时具
[13]有酸碱活性位,适当地改变镁铝比以及起中和作用的阴离子可以改变层板氧原子的电荷密度,从而调变这类催化剂表面酸碱活性位的比例。Dumitriu等制备了一系列不同镁铝比以及中和阴离子
的镁铝水滑石类催化剂,通过研究发现当镁铝比为3,中和阴离子为碳酸根时其碱性最强,催化环己醇脱氢生成环己酮的选择性可达到8619%。另外由于具有独特的层柱状结构,水滑石类催化剂的比表面通常比较高,可达到200mΠg。2
由上述可知,通过改变制备条件和选择不同的前驱体,可以制备具有强碱位甚至超强碱位的碱金属及碱土金属氧化物,但是这一类催化剂的比表面积都很小;水合滑石类催化剂虽然具有较大的比表面积,但其表面同时具有酸碱活性位,而且很难得到强的碱性位(相对于MgO)。为此人们提出了将碱金属和碱土金属氧化物及其盐类负载到多孔材料上的负载型无机固体碱。
3.3 负载型无机固体碱
?596?化学通报 2002年第9期 http:ΠΠhxtb.icas.ac.cn目前负载型固体碱的载体主要有三氧化铝和分子筛两种,此外也有用活性炭、氧化镁、氧化钙、二氧化锆、二氧化钛等作为载体的。负载的前驱体物种主要为碱金属、碱金属氢氧化物、碳酸盐、氟
[14~17]化物、硝酸盐、醋酸盐、氨化物和叠氮化物。由于碱土金属的氢氧化物和碳酸盐均为难溶物,
[18]而其硝酸盐分解温度普遍较高,所以也有少量报道用碱土金属醋酸盐作为前驱体的。至于负载
型无机固体碱的活性位主要是碱金属、碱金属氨化物、碱金属或碱土金属氧化物、氢氧化物、和碳酸盐,也有前驱体经高温煅烧后和载体反应生成的活性位。下面将结合负载前驱体和碱活性位的种类详细介绍不同载体的负载型固体碱催化剂。
31311 三氧化铝为载体的无机固体碱 三氧化铝表面同时具有酸碱活性位,其哈密特常数H-最高可以达到15。同时其机械强度高,热稳定性好是工业催化剂常用的载体。将上述的碱金属和碱土金属前驱体负载到三氧化铝表面,。
[16]由于氢氧化钾的碱性较强(H-=1814),,因此以钾的
K,将其负载到Al2O3表面,,KF和KNO3负载到Al2O3表面,。例如KF在高温焙
[19]烧过程中会和3KOH和KALO2类的碱物种:
12KF+Al2O3+3H2O
6KF+2Al2O32K3AlF6+6KOHK3AlF6+KALO2
[20]
[21][19][14~17][16](1)(2)KNO3和K2CO3的分解温度都比较高,分别为943K和1273K,但是当其负载在Al2O3表面
(3)时,在873K就可以和表面的羟基作用而产生AlKNO3+AlK2CO3+2AlOK活性位Al:OK(4)OKCO2+H2O+2Al
表1列出了氢氧化钾、碳酸钾以及一
些钾盐负载三氧化铝后经高温煅烧所得
[16,19]固体碱的碱强度。由表1可知,负载
钾盐或氢氧化物的三氧化铝经高温焙烧
后其碱强度顺序为KNO3ΠAl2O3=K2CO3Π
Al2O3>KFΠAl2O3>KOHΠAl2O3。表1 钾盐和碱以及负载在Al2O3上钾盐和碱的碱强度Tab.1 Basestrengthofpotassiumbaseandsaltaswellasthembeingloadedonaluminia固体碱K2CO3
KOH
13%KOHΠAl2O3
16%KFΠAl2O3
20%K2CO3ΠAl2O3
26%KNO3ΠAl2O3处理温度ΠK--773773873873H-151018141814>18142727此外,将强碱性的金属钠、氨基钾、或者CsOH等负载到Al2O3表面,经高温焙烧可以得到H->30的固体超强碱[22]。但是这些固体超强碱的前驱体碱性极强,
很容易被空气中的氧气、水等污染,并且
制备条件苛刻。采用弱碱性的Cs的碳酸
盐或者醋酸盐为前驱体通过浸渍法负载
到三氧化铝表面,高温分解后可以产生
H->35的超强碱位,如表2所示[22]表2 一些固体超强碱的碱强度Tab.2 Basestrengthofsomesupersolidbase固体碱
NaΠAl2O3
NaΠNaOHΠAl2O3
CsxOΠAl2O3前驱体和制备方法Na气相沉积Na、NaOH浸渍CsOAC浸渍处理温度ΠK823773973H-3535。由上述可知,以三氧化铝为载体制备固体碱的优点是:碱强度高(H-最高可达
37),机械强度和热稳定性好。其主要缺≥37
http:ΠΠhxtb.icas.ac.cn 化学通报 2002年第9期
2?597?点是:比表面积低,比表面为219mΠg的γ2Al2O3负载2mmolΠgCsOAC,经973K抽真空后比表面积降
到129mΠg2[22];此外三氧化铝的孔径分布较宽,分子择形性较差。
[19]31312 分子筛为载体的固体碱 沸石分子筛因其高比表面积和独特的择形性,而被广泛用作负载型固体碱的载体。按制备方法和碱活性位的种类来分,固体碱分子筛可以分为碱金属离子交换分子
筛、通过物理或化学方法将碱金属或稀土金属以金属态或金属氨化物形态分散到分子筛所得的固体碱(在此称为金属活性位固体碱分子筛)以及将弱碱性化合物作为碱位前躯体负载在高比表面沸石上再通过适当处理而产生强碱位所得的固体碱(在此称为弱碱位前躯体负载固体碱分子筛)。
(1)碱金属离子交换分子筛:硅铝分子筛中,以Si和Al为中心的SiO4和AlO4四面体通过共点、共边或共面构成具有一定孔结构的分子筛三维骨架。由于四配位的Al带有部分正电荷使得与其相邻的骨架氧因具有负电荷而呈碱性[23],。将分
[24]子筛与碱金属阳离子进行离子交换,增强并最终使分子筛呈不同强度的碱性。。但是通过碱金属离子交换制备的固、骨架氧电负性最强的X型分子筛经过Cs交换,在423K1015%。
(2)金属活性位固体碱分子筛:通过碱金属气相沉积、负载碱金属重氮盐、有机碱金属溶液以及钠Π液氨浸渍[19][25]等方法可以在沸石表面形成类似Na43+的碱金属簇活性位,从而制备固体超强碱。此外,Baba等将镧系金属Yb或Eu溶于液氨后,负载在NaY、NaX、NaM、NaA、NaZSM25沸石上形成了EuNHx(x=1,2)物类和金属微粒,制备了强碱性沸石催化剂,在需要强碱位催化的丁烯异构化反应中具有较高活性。但由于这些固体超强碱的活性位主要为金属簇或金属氨化物,很容易被空气中水、氧气和二氧化碳中毒而难以工业化应用。
(3)弱碱位或中性前躯体负载固体碱分子筛:在沸石分子筛表面引入碱性客体,使其在高温活化过程中产生类似于碱金属氢氧化物或氧化物的活性位也可以制备强碱性沸石分子筛。但是如果在沸石上直接负载具有强碱性的碱金属氧化物或氢氧化物,固然形成了强的碱性位,可是在催化剂的高温活化过程中这些碱金属氧化物或氢氧化物,会和分子筛中的硅组分反应,侵蚀分子筛的骨架而造成分子筛的结构坍塌。将弱碱性或中性化合物作为碱位前躯体负载在高比表面沸石上再通过适当处理而产生强碱位,能避免对于沸石结构的破坏,从而成为制备强碱性沸石的另一重要途径。目前常用Cs盐和钾盐水溶液浸渍沸石,然后加热分解而生成碱金属氧化物簇,在需要弱碱位或中强碱位催化的的异丙醇脱氢和需要强碱位催化的丁烯异构化反应中都具有较高活性[27][26]。
[28]由于在碱金属元素中以Cs的碱性最强,并且用CsOAc改性Al2O3可以制备出H-高于30的固体超强碱。因此Cs盐被尝试着用来改性分子筛从而制备强碱性固体碱分子筛。Meyer等用
CsOAc改性的CsX分子筛催化苯甲酸甲酯和乙二醇的酯交换反应中,其转化率和选择性分别达到了8210%和9017%。随着MCM241中孔分子筛的出现,碱性分子筛的种类又有所增加。Kloestra等[29]将CsOAc负载在MCM241,得到孔径约为3nm的强碱性中孔分子筛;在423K的丙二酸二乙酯Michael加成反应中,其转化率和选择性分别达到了87%和92%。然而CsOAc分解产生的CsO会和分子筛载体骨架反应生成弱碱性硅酸盐,这一方面降低了其表面碱性,例如CsOΠCsX的碱强度为
[28](1712≤H-≤1814);另一方面使分子筛的比表面积急剧减小,以CsOAc改性MCM241时,当Cs负
载量高于22(wt)%之后,样品比表面积由1069mΠg降到176mΠg22[29]。
此外,由于用KF、KNO3、K2CO3改性Al2O3可以得到H-=1814~27的固体碱,而且弱碱性或中
?598?化学通报 2002年第9期 http:ΠΠhxtb.icas.ac.cn性的钾盐负载在分子筛表面,不会侵蚀分子筛的骨架,因此钾盐改性的分子筛成为固体碱分子筛研究领域的热点。将KF负载在NaY分子筛上,KF会和沸石中的硅铝组分发生类似于和Al2O3、SiO2的相互作用而产生KOH类的碱物种[19]。但是KFΠNaY的碱强度却很低(H-<>
6KOH+2K3AlF6
K2SiF6+4KOH
[30]12KF+3H2O+Al2O36KF+SiO2+2H2O(5)(6)将KNO3负载在Al2O3表面873K就能分解产生Al—OK超强碱位,但是将其负载在NaY分子筛上却很难分解,21(wt)%KNO3ΠNaY经过873K抽空后只产生0116mmolΠg的碱量
0195mmolΠg的碱量,碱强度达到H-=1510[30],用异丙醇、丙酮、乙醇等还原剂在673K处理21(wt)%KNO3ΠNaY,则可以通过氧化还原过程分解KNO3而产生。由上述可知,在NaY分子筛上用KF和KNO3改性都得不到类似于三氧化铝改性的固体强碱。这主要是因为NaY钾化物生成碱强度低的硅钾化合物,塌,例如16(wt)%KFΠNaY经923K将NaY分子筛用Al2,碱强度,。Al2O3可以通过化学沉积[31][19]。用KF改性的Al2O3ΠNaY碱强度可以达到H-=1712[19],而用KNO3改性的Al2O3ΠNaY,在873K就可以使KNO3分解,并且产生较高的碱强度(H-=1712~1814)。
除了对分子筛进行表面镀饰外,选择适当结构的分子筛进行钾盐改性也能得到强碱性分子筛,例如将KF负载在与NaY分子筛结构不同但硅铝比相近的KL分子筛上其碱强度能达到H-=
[19]1510;将KNO3和K2CO3负载在KL上,873K抽空KNO3和K2CO3就会分解而产生具有超强碱位
[33]的KO微粒(H-=2710)β。然而除了KL之外的各种沸石如NaY、KNaZSM25、KY和KX负载了
。此外KL沸石的比表面积较小(246mΠg)2[33]KNO3后都没有超强碱位[19]而且KL沸石改性需要负
载10%~20%的KNO3或K2CO3才能出现超强碱位,其孔道被严重阻塞以至于正己烷吸附量(298K,p/ps=015)减少50%~80%[33]。
由于碱土金属碳酸盐难溶于水,而且其硝酸盐分解温度高,所以很难将碱土金属以盐的形式负
[34]载到分子筛表面。朱建华等将Mg(OH)2高温煅烧所得MgO经微波辐射法负载在NaY和KL分
子筛上得到了碱强度高于MgO(H-≤1814)的碱性位(H-=2215)。与KF或KNO3改性法相比,MgO在微波辐射条件下既不会破坏沸石结构,也不产生MgAl2O4之类的新物象,此外该方法对沸石孔结构的影响较小。KL沸石负载10(wt)%的MgO后,其苯吸附量仅仅减少12%。使用微波辐射在沸石上负载MgO还具有耗时少、操作简单等特点,为研制强碱性分子筛开辟了新途径。
31313 其它载体的固体碱 除了被广泛使用的Al2O3和分子筛外,活性炭、ZrO2、TiO2、MgO、CaO也可以作为负载型固体碱的载体。相同的前驱体负载在不同载体上其碱强度随载体碱强度的增大而
[35]增大。此外,将氢氧化钠负载在强碱性的MgO或CaO上823K煅烧甚至可以制备H->2615的固体超强碱。但是这些载体除活性炭外,比表面积都比较小且机械强度低。
总而言之,将碱金属或碱金属、碱土金属氧化物及其盐和碱负载到多孔载体上,不仅可以得到超强碱位的固体碱而且还可以显著提高催化剂的比表面积,是制备固体碱催化剂的主要研究方向。[36]4 展望
综上所述,固体碱种类繁多,覆盖的碱强度范围宽,因此固体碱的选择和研究方向应视其所应
http:ΠΠhxtb.icas.ac.cn 化学通报 2002年第9期?599?用的化学反应而定。
(1)对于需要弱碱位催化的反应,例如Knoevenagel和羟醛缩合反应,可以选用有机碱负载的分子筛以及碱金属离子交换的分子筛。这一类固体碱催化剂的优点是比表面积大、分子择形性好、选择不同的分子筛还可以控制载体的孔径。此外有机碱负载的分子筛碱强度均匀,且活性组分不流失;而碱金属交换分子筛制备简单,可以适用于高温反应。这一类固体碱需要提高的是其机械强度,尤其在适用于液相反应的时候。
(2)对于需要中强碱和强碱位催化的反应,例如异丙醇脱氢和丁烯异构,可以选用钾盐改性的三氧化铝和分子筛。这一类催化剂具有较高的碱强度;且三氧化铝改性的固体碱碱强度高,热稳定性好;而分子筛改性的催化剂比表面积高,分子择形性好。这一类催化剂需要改进的是在保证碱强度的同时,提高其比表面积和机械强度。
(3)对于需要超强碱位催化的反应,例如12炔和醛铝。这一类催化剂碱强度高,但是成本高,,和降低成本入手。
(4),。对于高比表面积、,应重视其碱强度、结构稳定性和机械强度的提高;而对于碱强;此外也可以考虑用高比表面积的活性炭作为载体,其研究的重点应为机械强度、碱强度以及孔径分布的控制;而其它载体由于比表面积低且机械强度差,所以工业应用价值较小。
参考文献
[1] ChoudaryBM,KantamML,SanthiP.Catal.Today,2000,57:17~32.
[2] BrunelD.MicroporousandMesoporousMaterials,1999,27:329~344.
[3] DoskocilEJ,BordawekarSV,DavisRJ.J.Catal.,1997,169:327~337.
[4] ZhangG,HattoriH,TanabeK.Appl.Catal.,1988,36:189~197.
[5] PinesH,VeselyJA,IpatieffVN.J.Am.Chem.Soc.,1955,77:347~348.
[6] PachecoMA,MarshallCL.Energy&Fuels,1997,11:2~29.
[7] 东北师范大学,华南师范大学等编.有机化学(下册).北京:高等教育出版社,1992:88~89.
[8] LinX,ChuahGK,JaenickeS.J.Mol.Catal.A:Chemical,1999,150:287~294.
[9] CollucciaS,BoccuzziF,GhiottiGetal.J.Am.Chem.Soc.,FaradayTrans.,1982,78(1):2111~2119.
[10] TanabeK,MisonoM,OnoYetal.StudiesintheSurfaceScienceandCatalysis(Vol.51).Amsterdam:Elsevier,1989:27~30.
[11] KabashimaH,ShibuyaHT,HattoriH.J.Mol.Catal.A:Chemical,2000,155:23~29.
[12] 胡长文,李丹峰,郭伊荇等.科学通报,2001,46(5):359~364.
[13] DumitriuE,HuleaV,ChelaruCetal.Appl.Catal.A:General,1999,178:145~157.
[14] TanabeK,HolderichWF.Appl.Catal.A:General,1999,181:399~434.
[15] FuY,BabaT,OnoY.Appl.Calal.A:General,1998,166:425~430.
[16] ZhuJH,WangY,ChunYetal.J.Am.Chem.Soc.,FaradayTrans.,1998,94:1163~1169.
[17] HandaH,BabaT,OnoY.J.Chem.Soc.FaradayTrans.,1998,94:451~454.
[18] BordawejarSV,DosjocilEJ,DavisRJ.Langmuir,1998,14:1734~1738.
[19] 朱建华,淳 远,王 英等.科学通报,1999,44:897~902.
[20] 刘预知.无机物质理化性质及重要反应方程式手册.成都:成都科技大学出版社,1993:358~453.
[21] 朱建华,王 英.催化学报,1996,17:286~290.
[22] GozawsjiH,HoelderichWF.Appl.Catal.A:General,1999,179:131~137.
[23] WielandWS,DavisRJ,GarcesJM.Catal.,1998,173:490~500.
[24] TanabeK,MisonoM,OnoYetal.StudiesintheSurfaceScienceandCatalysis(Vol.51).Amsterdam:Elsevier,1989:150~151.
?600?化学通报 2002年第9期 http:ΠΠhxtb.icas.ac.cn
[25] DoskocilEJ,BordawekarSV,KayeBGetal.J.Phys.Chem.B,1999,103:6277~6282.
[26] BabaT,KimGJ,OnoY.J.Am.Chem.Soc.,FaradayTrans.,1992,88(6):891~897.
[27] HathawayPE,DavisME.J.Catal.,1989,116:279~284.
[28] MeyerU,HoelderichWF.Appl.Catal.A:General,1999,178:159~166.
[29] KloetstraKR,BekkumHV.Stud.Surf.Sci.Catal.,1997,105:431~438.
[30] 淳 远,朱建华,须沁华.科学通报,1996,41:1858~1860.
[31] 朱建华,淳 远,须沁华等.科学通报,1997,42:112.
[32] 朱建华,孙德坤,时 吉等.科学通报,1998,43;109~110.
[33] 淳 远,朱建华,王 英等.科学通报,1998,43:721~724.
[34] 王 英,淳 远,朱建华等.催化学报,1999,20:409~414.[35] BordawekarSV,DoskocilEJ,DavisRJ.Catal.Lett.,1997,44:193~199.
[36] RuckensteinE,KhanAZ.J.Catal.,1993,141:628~647.?信息服务?
—
彩页及DEMO盘,代理世界主要化学软件产品,并可为。我公司代理产品如下:
最著名的化学软件公司
11CambridgeSoftCorp 主要产品,功能特征为科研用户特惠价格
www.camsoft.com
21GaussianInc
www.Gaussian.com
3.HypercubeInc
www.hyper.com
4.SigmsoftCorp
www.sigmasoftc.com
5.ChemSW,Inc
www.chemsw.com
6.SciVisionInc
www.scivision.com
7.Cachesoftware
www.cachesoftware.com
8.ModelSciSoft
http:ΠΠmodelscience.com
WardsysΠXoΠBirminghamChemOfficeUltra2002,ChemDrawChem3D,ChemFinder等一系列完整的软件ChemOfficeUltra200211500元(学生6000元)G98W7000元GVW6000元G9822500元Hyperchem71011500元Atlantis20001550元MolSuiteTM20005400元TM著名的量子化学计算软件,采用非经验分子轨道方法,计算分子的立体结构。计算精度高,用户可以根据要求的计算时间选用不同的计算精度分子模拟软件。3D建模动画、可提供量子化学、分子结构、分子动力学等方面的计算数学图形和计算软件是现今唯一的为能解决工业生产和数学应用而设计的图形计算软件产品MolSuite2000为您提供分子模型图库,物理属性计算,统计分析,以及数据库开发管理。生物材料环境化学家们不可多得的帮手,包含大量固体光学常数以及各类有机、无机合物的红外、拉曼光谱FUJITSU公司的最新化学软件产品CacheworksystemproMopac各版本2000~35000元各版本2000~26500元3000元最有用的化学实验软件ModelChemLabGeneHunterΠHASLpcv3.30ΠMolrpo价格优惠 地址:北京海淀体育中心科研楼403、404室 邮编:100080
Tel:010262563892 Fax:010262526467 网址:www.hongcam.com E2mail:sales@hongcam.com
范文二:固体碱催化剂
固体碱催化剂
1 引言
固体碱指能向反应物给出电子的固体物质,也可以说是能够化学吸附酸的固体物质或能使酸性指示剂变色的固体物质。固体碱作为催化剂,具有活性高、选择性高、反应条件温和、易与产物分离、易于再生等诸多的优点。另外,固体碱催化剂对反应设备的腐蚀性较小,延长了设备的使用寿命,增强了设备的生产能力。在精细化工中,固体碱催化剂可以提高反应的选择性、提高产品的转化率、降低能耗和废物的排放量。
固体碱催化剂的研究发展较慢、缺乏系统性,这主要是由于固体碱催化剂制备复杂、成本较高、强度较差、易被水和二氧化碳等污染而中毒、并且催化剂比表面积较小。近些年来,越来越多的人都正在积极地研究如何制备应用性较强的固体碱催化剂。目前,全球都在提倡绿色化工和对环境有好的化工,因此,固体碱催化剂具有极高的研究意义。
2 固体碱的分类
按照不同的分类方法,可以将固体碱催化剂分为不同的催化体系。本文将固体碱分为有机固体碱、有机无机复合固体碱、阴离子交换树脂和无机固体碱,其中又将无机固体碱分为金属氧化物型固体碱、金属含氧酸盐型固体碱和负载型固体碱。
2.1 有机固体碱
有机固体碱一般是指结构末端为叔胺或叔膦基团的碱性树脂类固体碱,但是按照广义的酸碱理论来说,醇的碱金属盐类、烷基金属锂化物和格氏试剂等都是有机固体碱。虽然有机固体碱得碱强度均一,但是其不适合用于工业生产中,因为有机固体碱的热稳定性不好,高温下分解,只能用于低温反应中,另外,有机固体碱的制备比较复杂,制备费用昂贵也是其发展前景不被看好的原因。
2.2 有机无机复合固体碱
有机无机复合固体碱是一种负载型固体碱,主要是将有机胺和季铵碱负载在分子筛上。有机无机复合固体碱的碱强度均匀,并且活性组分不会流失。但是由于其活性位是有机碱,因此,有机无机复合固体碱与有机固体碱一样,不能应用于高温反应中,制备也比较困难。
2.3 阴离子交换树脂
阴离子交换树脂一般呈现多孔状或颗粒状,大小约为0.1~1mm,在水或极性溶剂中发生溶胀,在水中具有碱性,能以其氢氧根离子或非金属离子交换溶液中的阴离子。阴离子交换树脂是近年来新兴的碱性催化剂。根据离子的交换能力可以将阴离子交换树脂分为强碱型阴离子交换树脂、弱碱型阴离子交换树脂和对阴离子的吸附。
阴离子交换树脂可应用于羟醛缩合反应。欧植泽等通过试验筛选出三丁胺胺化强碱性阴离子交换树脂作为相转移催化剂,催化合成苄叉丙酮,在优化反应条件下,苄叉丙酮的收率可达98%,且催化剂可重复使用。胡微等选择强碱性苯乙烯系季铵型离子交换树脂作为催化剂,用丙酮与甲醛缩合制得乙酰乙醇,再在草酸存在下脱水得到甲基乙烯酮。石秀敏等研制
筛选出适合于催化蒸馏法制二丙酮醇的新型阴离子催化剂,即大孔型强碱性苯乙烯阴离子树脂,催化活性和选择性都很高。
2.4 无机固体碱
无机固体碱是固体碱研究的主要领域,它代表着固体碱的研究发展方向。相对于其它种类固体碱,无机固体碱的特点是制备简单、碱强度分布范围宽、热稳定性好,具有非常广阔的研究前景。无机固体碱主要分为:金属氧化物型固体碱、金属含氧酸盐型固体碱和负载型固体碱。
2.4.1 金属氧化物型固体碱
金属氧化物型固体碱可以分为单一金属氧化物型固体碱、复合金属氧化物型固体碱和稀土金属氧化物型固体碱。
2.4.1.1 单一金属氧化物型固体碱
单一金属氧化物型固体碱是研究开发比较早的一类固体碱,它主要包括碱金属氧化物和碱土金属氧化物,这一类固体碱的碱性位主要来源于表面吸附水后产生的羟基和带负电的晶格氧。目前,有关单一金属氧化物型固体碱的研究报道主要集中在MgO、CaO、BaO、KO、2SrO等物质,但主要还是以MgO为主的研究比较多。氧化镁固体碱的制备方法主要有:将MgO负载在AlO、ZrO和各种分子筛等载体上;以硝酸盐等物质作为前躯体加热分解得到。 232
一般的,碱金属和碱土金属氧化物催化剂的碱性强度随着原子序数的增加而增加,即:NaO<><>
小。影响碱金属氧化物和碱土金属氧化物碱强度的主要因素是煅烧温度和前躯体。一般的,较高的煅烧温度可以获得较强的碱性位,通过不同前躯体所获得得氧化物的碱性强度顺序为:碳酸盐>氢氧化物>醋酸盐。但是这类固体碱均呈现为粉末状,没有机械强度,不易从产物中分离,另外,除MgO外,其余的比表面积都较小。诸多的缺点阻碍着此类固体碱的研究和发展。
2.4.1.2 复合金属氧化物型固体碱
复合金属氧化物型固体碱又称为水合滑石类固体碱,这类固体碱是一种中孔材料,一
2+3+m-般是通过前驱体煅烧制得,前驱体结构式为:[MM(OH)](A)?nHO,其中,M122(x+1)1/m21
m-和M均为金属阳离子,A为阴离子。复合金属氧化物型固体碱的比表面积大、稳定性好,21/m
但是其表面同时具有酸碱活性位,很难得到强碱性复合金属氧化物型固体碱。
2.4.1.3 稀土金属氧化物型固体碱
稀土金属氧化物在一些反应中作为催化剂和助催化剂时具有良好的催化活性和选择性。稀土金属氧化物的碱性较强,这主要是由于其阳离子半径较大、氧配位数较多和氧的供电子能力较强。600?以下时,稀土金属氧化物表面存在强弱两种固体碱活性中心,其一是强碱中心,其碱强度比MgO表面的强碱中心强得多,碱强度顺序是LaO>NdO>YO>CeO>MgO;其二是弱碱中心,其碱强度与MgO弱碱中心强度差不多,2323232
碱强度顺序是:NdO>ZrO> YO> MgO> CeO> LaO。 23223223
2.4.2 金属含氧酸盐型固体碱
金属含氧酸盐型固体碱主要有NaCO、KCO、KHCO、KNO等,它们可以单独作为催232333
化剂使用,但是由于碱量和碱强度不太理想,因此,多把金属含氧酸盐作为负载型固体碱的前驱体来进行使用。
2.4.3 负载型固体碱
目前,负载型固体碱的载体主要有AlO、分子筛和ZrO等,另外,也有用MgO、CaO、232
TiO和活性炭等物质作为载体的。负载的前驱体主要有碱金属、碱金属氧化物、碱金属氢氧2
化物、碳酸盐、氟化物、硝酸盐、醋酸盐、氨化物和叠氮化合物。负载型固体碱的活性位主要有碱金属、碱金属氨化物、碱金属及碱土金属氧化物、氢氧化物、碳酸盐,也有前驱体经高温煅烧后和载体反应生成的活性位。
2.4.3.1 AlO为载体的固体碱 23
AlO是工业催化剂常用的载体。将前驱体负载到AlO表面,经高温煅烧可得到不同碱2323
强度的负载型固体碱。目前,以AlO为载体的固体碱研究较多的是负载钾盐或氢氧化钾。23
KOH和KCO本身均具有碱性,将其负载到AlO表面,前驱体本身就可以产生活性位;而2323
中性盐KF和KNO等负载到AlO表面需要经过高温煅烧才能获得碱性位。以AlO为载体32323制备的固体碱的优点是碱强度高(H最高可以达到37)、较高的机械强度和较好的热稳定性。-
而其缺点是比表面积较低,AlO孔径较宽导致分子选择性较差。 23
2.4.3.2 分子筛为载体的固体碱
分子筛固体碱被选择作为负载型固体碱的载体,这主要是因为其具有高比表面积和独特的择形性。按制备方法和碱活性位的种类可以将以分子筛为载体的固体碱分为碱金属离子交换分子筛、金属活性位固体碱分子筛和弱碱位或中性前驱体负载固体碱分子筛。
(1) 碱金属离子交换分子筛
分子筛与碱金属阳离子进行离子交换的过程中,使金属阳离子进入分子筛的笼中,导致骨架氧的电负性增强并最终使分子筛呈现不同强度的碱性。碱金属交换所得分子筛的碱性随着硅铝比的减小以及交换金属阳离子半径的增大而增加。但是,通过离子交换制备的固体碱分子筛只能产生弱碱位。
(2) 金属活性位固体碱分子筛
金属活性位固体碱分子筛是通过物理或化学方法将碱金属或稀土金属以金属态或金属氨化物形态分散在分子筛所得的固体碱。但是,该类固体碱的活性位主要为金属簇或金属氨化物,很容易被空气中的水分、氧气和二氧化碳中毒而难以得到工业化应用。 (3) 弱碱位或中性前驱体负载固体碱分子筛
该类固体碱是将弱碱性化合物作为碱位前驱体负载在高比表面沸石上再通过适当处理而产生强碱位而得到,这一途径可以避免分子筛的结构遭到破坏。目前,常用的该类固体碱
是用铯盐和钾盐水溶液浸渍沸石,然后加热分解而生成碱金属氧化物簇。
2.4.3.3 ZrO为载体的固体碱 2
目前,有关以ZrO为载体固体碱的研究报道较少,但是其将在未来研究中充满竞争力,2
因为ZrO表面同时具有酸位和碱位及氧化-还原性质可作为优良的新型载体。一般可以将2
KNO、KCO、Mg(NO)、MgO等碱金属或碱土金属的氧化物及盐类负载在ZrO的表面来制323322备此类固体碱,ZrO为载体的固体碱的碱强度可以达到较高强度。但美中不足的是其比表面2
积较小,机械强度较低。
2.4.3.4 其它载体的固体碱
负载型固体碱的载体除了以上三种外,也可用活性炭、MgO、CaO、TiO作为载体。相2同的前驱体负载在不同的载体上,其碱强度随着载体减轻度的增大而增大。但是,除了活性炭外,其他类载体的比表面积较小、机械强度低及易受HO和CO等杂质污染。 22
通过改变前驱体和采用不同的方法可以制备具有不同碱位、不同碱强度的固体碱,从而使制得的固体碱满足不同反应的需要。将碱金属或碱金属、碱土金属氧化物及其碱和盐负载到多孔载体上,不但可以获得较高强度的固体碱,还可以显著提高固体碱的表面积。制备多孔载体固体碱是固体碱催化剂的主要研究方向。
3 固体碱催化剂在有机合成反应中的应用举例
3.1 异构化反应
1-丁烯异构化生成2-丁烯常被用来表征催化剂的性能。AlO、MgO和CaO对此反应均23
具有较好的催化效果。反应过程中,催化剂的碱中心首先吸收烯丙基上的氢,形成顺式和反式的烯丙基离子,由于顺式比反式的烯丙基稳定,因此通过固体碱催化剂可以控制最终产物主要为顺式异构化产物。固体碱催化剂对于含有氧或氮等杂原子的烯烃化合物异构化反应具有显著的优势,因为酸性催化剂容易与杂原子相互作用使催化剂中毒失去活性,而固体碱催化剂可以避免这种影响,达到良好的催化效果。
3.2 氧化反应
在环戊烯酮氧化反应过程中,TBHP-KF/AlO催化剂表现出了优良的催化性能,其中TBHP23
是指叔丁基过氧化氢。反应过程中,氧化剂在固体碱催化剂的作用下释放出H质子变为亲核试剂,从而与不饱和烯烃发生环氧化反应。反应式如下:
(1)
3.3 氨化反应
伯胺和仲胺在固体碱的催化作用下能够与共轭双烯发生加成反应。碱土金属氧化物、
LaO和ThO都表现出了较高的活性。反应方程式如下: 232
(2)
--+反应过程中,胺在固体碱的作用下形成R(R)N,然后R(R)N和H便顺利的与烯烃发1212
生加成反应。
3.4 氢化反应
+-与氨化反应一样,氢分子在固体碱的作用下形成H和H,然后再发生加成反应。例如,在0?下,1,3-丁二烯在固体碱MgO的作用下发生氢化反应,反应方程式如下:
(3)
3.5 C-C键合成反应
3.5.1 醇醛缩合反应
研究表明:固体碱金属氧化物在丙酮自身缩合反应中表现出了良好的催化性能。另外,60?时,丙酮与苯甲醛在复合金属氧化物型固体碱的催化作用下可以发生缩合反应反应式如下:
(4)
3.5.2 Knoevenagel缩合反应
Knoevenagel缩合反应发生在酮和含活性亚甲基的化合物之间,常用的固体碱催化剂有碱性离子交换的沸石、海泡石、氮氧化物、二甲胺乙基磷酸钡和负载有KF的磷酸盐。在反应的过程中,固体碱催化剂均表现出了较高的活性并且容易分离和再循环利用,另外,反应条件温和。
3.5.3 Nitroaldol反应
含羰基的化合物与含硝基的烷烃发生的加成反应被称作Nitroaldol反应。反应式如下:
(5)
在Nitroaldol反应中,碱性氧化物、氢氧化物和KF/AlO都对此反应具有催化活性。以23
+硝基甲烷与丙醛反应生成1-硝基-2丁醇为例,硝基甲烷在碱性催化剂的作用下失去H变成
+阴离子,然后阴离子进攻羰基碳,最后结合H得到最终产物。在反应过程中,固体碱催化剂不易被空气污染,主要是因为硝基甲烷强吸附在催化剂表面并且吸附了的CO可以被硝基2甲烷取代。
3.5.4 Michael加成反应
Michael加成反应多发生在含有活性亚甲基化合物与含有α、β不饱和羰基化合物之间。例子如下:
(6)
在反应的过程中,KF/AlO和KOH/ AlO均具有较高的催化活性,产物的选择性较高。2323
这种催化剂相比于液态碱性催化剂具有较大的优势。
3.5.5 醇的加成反应
醇与不饱和酮的反应中,通常选用碱土金属氧化物、KF/AlO和KOH/ AlO作为催化剂,2323它们均具有价高的催化活性。反应通式如下:
(7)
但对于甲醇的加成反应,MgO和CaO均显示出了较高的催化活性。在甲醇的加成反应中,甲醇在催化剂上较强的吸附作用使得催化剂暴漏在空气中也不会被污染。
3.5.6 苯基炔烃亲核加成反应
苯基炔烃亲核加成反应通常以KNH/ AlO为催化剂,可以显著地提高产物选择性和收223
+率。反应过程是苯基炔烃中三键上的氢在固体碱催化剂的作用下失去H变为负电荷的阴离子,此后,阴离子进攻另一反应物,最终得到产物。
3.5.7 Wadsworth—Emmons反应
传统的方法是用液体碱催化剂来催化该反应的进行,KF/AlO、MgO和ZnO也能催化该23
t反应,但是催化效果一般。目前,具有较高催化活性的是Mg—Al—水滑石—OBu,可以通过改变镁铝比例来调节催化性能。反应式如下:
(8)
3.6 Si—C键合成反应
该类反应通常以KNH/ AlO或KF/AlO为催化剂,例子如下: 22323
(9)
+反应机理是MeSiC—CH在固体碱催化剂的作用下失去H被固体碱吸附,从而变为3
—MeSiC—C,得到的阴离子进攻另一反应分子中的Si,从而形成另一含有Si—C键的化合物。 3
3.7 P—C键合成反应
Pudovic反应是含有一个不稳定的P—C键的化合物与另一不饱和化合物之间的加成反应,在该反应中,KOH/ AlO具有较高的催化活性,可以使产物的选择性和收率均得到较大23
提高。
3.8 环的反应
3.8.1 开环反应
脱水甘油与脂肪酸制备甘油酸酯是典型的开环反应,孔分子筛MCM—41和MAF固体碱催化剂(经过热水处理的镁铝复合氧化物浸渍适量的KF)具有显著的催化效果,并且具有选择性高、用后无需处理、可以重复使用等优势。
3.8.2 杂环合成反应
以4—甲基噻唑的合成反应为例,反应式如下:
(10)
在该反应中,负载有Cs的ZSM—5沸石具有较理想的活性、选择性和寿命。
4 小结
目前,固体碱催化剂的研究还不够系统和深入,仅研究了固体碱催化剂在部分反应中的应用,其实,还有很多反应可以用固体碱催化剂。固体碱催化剂在工业中应用可以满足清洁生产和环保的要求,它将是催化剂领域发展的必然趋势,并且会推动有机合成的发展。
固体碱催化剂的种类繁多,碱强度范围宽,因此有关固体碱催化剂的选择要根据所应用的化学反应来确定。
(1) 弱碱位催化反应
对于这类反应可以选用有机碱负载的分子筛及碱金属离子交换的分子筛。这类固体碱催化剂的比表面积大,分子择形性好,选择不同的分子筛还可以控制载体的孔径。此外有机碱负载的分子筛碱强度均匀,且活性组分不流失;而碱金属交换分子筛制备简单,可以适用于高温反应。这一类固体碱需要提高的是其机械强度,尤其在适用于液相反应的时候。Knoevenagel和羟醛缩合反应均属于这类反应。
(2) 中强碱和强碱位催化反应
在这类反应中,可以选用钾盐改性的三氧化铝和分子筛。这一类催化剂具有较高的碱强度;且三氧化铝改性的固体碱碱强度高,热稳定性好,而而分子筛改性的催化剂比表面积高,分子择形性好。这一类催化剂需要改进的是在保证碱强度的同时,提高其比表面积和机械强度。异丙醇脱氢和丁烯异构均属于这类反应。
(3) 超强碱位催化反应
在类反应中,可以选用Cs改性的三氧化铝。这一类催化剂碱强度高,但是成本高,比表面积低,因此在研制开发时应主要从提高比表面积和降低成本入手。1-炔和醛、酮的缩合反应属于这类反应。
不同载体的负载型固体碱催化剂,其未来研究的侧重点也应有所不同。对于比表面积高、分子择形性好的改性分子筛固体碱,应重视提高其碱强度、结构稳定性和机械强度;而对于碱强度较高的改性三氧化铝应重视提高载体比表面积和控制孔径分布;此外也可以考虑用高比表面积的活性炭作为载体,其研究的重点应为控制机械强度、碱强度以及孔径分布;而其它载体由于比表面积低且机械强度差,所以工业应用价值较小。
固体超强碱催化剂
1 引言
-固体超强碱是指常温下碱强度(哈密特常数H= lg([A]/[HA])+PK)大于26的物质,也可_BH
以认为是碱强度(H=7)比中性物质高出19个单位的物质。近年来,越来越多的人致力于研0
究固体超强碱作为催化剂在工业中的应用。固体超强碱催化剂有如下一些基本特征:(1) 固体超强碱催化剂克服了固体碱催化剂易被空气中的HO和CO污染的缺陷;(2) 固体超强碱22
催化剂的活性极高、可以使反应条件温和、可以在高温反应中使用;(3) 易与产物分离,使工艺得到简化,产物的纯度和选择性极高;(4) 催化剂可以循环使用,并可以连续使用;(5) 对反应设备腐蚀较小,并且产生的废水较少。
2 固体超强碱催化剂的种类与制备
目前,发现的固体超强碱催化剂主要有:碱金属氧化物、碱土金属氧化物及其氢氧化物、负载型碱金属及碱金属氢氧化物、中性盐负载在AlO上的超强碱。固体超强碱催化剂23
的制备方法可以分为两类:一类为非负载型的,一般由盐或碱经高温处理得到,如CaO、SrO;另一类为负载型的,以γ-AlO为载体,经强碱溶液浸渍灼烧后添加碱金属元素,如 γ23
-AlO-NaOH-Na。这些固体超强碱催化剂及制备方法如表1。 23
表1 固体超强碱催化剂及制备方法
3 有机合成实例中的固体超强碱催化剂
3.1 KNO/AlO型固体超强碱催化合成香豆素 323
一直以来,国内外对于香豆素的合成都是采用Perkin(铂金)法,即以水杨醛和醋酐为原料,醋酸钾为催化剂,这样得到的收率为70%左右,另外,副反应多,产品质量不稳定,能源消耗大。人们对香豆素的合成工艺不断地改进,据报道最高收率为80.6%。随着固体超强碱催化剂的出现,人们研究出了以KNO/AlO型固体超强碱为缩合催化剂的工艺,此工艺323
可以使香豆素的收率提高至85%以上,并且催化剂易于回收,减少了环境污染,产品符合标准。
KNO/AlO型固体超强碱催化剂的制备方法:(1) γ-AlO的制备:称取48 g异丙醇铝32323
于250mL圆底烧瓶中,加入168 mL异丙醇和18 mL水(水和异丙醇组成共沸物,共沸点为80.3?),于85?恒温进行反应,不断搅拌,4 h后停止反应,得白色糊状物。搭好蒸馏装置,将水及异丙醇蒸出,馏程为81~94?,得白色细粉状氢氧化铝。马弗炉中450?焙烧2 h,得γ-AlO载体。(2) 固体超强碱的制备:将质量比为26%的KNO与74%的γ-AlO混合,研磨,23323加入适量蒸馏水,研磨为糊状,110?烘干。在600?下焙烧5 h,即得到KNO/AlO型固体323超强碱。
KNO/AlO型固体超强碱催化剂具有的碱强度较高、热稳定性好,另外,比表面积可以323
2达到431m/g,碱强度H>26.5。 —
3.2 Na-NaOH-γ-AlO催化合成查尔酮 23
查尔酮传统经典的合成方法是苯乙酮与苯甲醛在强碱如醇钠或强酸的催化作用下进行交叉羟醛缩合而成,但是此方法的收率仅在30%~70%之间,并且对设备的腐蚀性较大、环境污染较严重、产物不易分离。另外,有文献报道采用烷基锂、芳基锡、NaOH和1-丁基-3-甲基六氟磷酸咪唑盐以及KF- AlO等作催化剂制备查尔酮,但结果都不理想,主要是这些23
催化剂制备困难、价格昂贵,并且催化反应时间长、催化效率较低。若以Na-NaOH-γ-AlO23催化合成查尔酮,反应在40?进行3h可以使收率达到近97%。另外,Na-NaOH-γ-AlO23还可应用于烯烃的双键转移反应、含杂原子不饱和化合物双键转移反应、共轭双烯的加氢加胺反应、烷基苯的侧链烷基化反应、酯化及酯交换反应。
Na-NaOH-γ-AlO固体碱催化剂的制备方法:在装有搅拌器反应釜中,加入粉状γ-AlO2323载体,开动搅拌器,N流环境中缓慢加热升温至250~350?,保温30~40min,待脱水完全2
后,加入15~40g的NaOH,继续搅拌2~3 h,然后在同一温度下分批加入6~10g的Na,继续反应1 h,得到蓝色或淡蓝色粉末状就是Na-NaOH-γ-AlO型固体超强碱。 23
Na-NaOH-γ-AlO固体碱的碱强度可以达到37以上,在其催化体系中,催化剂选择性23
高、反应条件温和、产物易于分离、生产清洁、可循环利用。另外,Na-NaOH-γ-AlO固23体碱对于环己酮羟醛缩合、烷基苯侧链烷基化和烯烃的双键异构化等的反应均有明显的效果。
3.3 NaCO/γ-AlO和NaOH/γ-AlO催化合成3-羟甲基乙烷 232323
三羟甲基乙烷通常由丙醛与甲醛经羟醛缩合反应首先生成2,2-二羟甲基丙醛,然后再与
过量甲醛在催化剂作用下经Cannizzaro反应而制得。羟醛缩合反应和Cannizzaro反应的催化剂主要有无机碱和有机碱等均相催化剂,由于它们都会溶解在溶液中,因此对最后的产物分离会造成很大困扰。而多相催化剂可以减少环境污染、降低设备的腐蚀、易于分离等,其中,NaCO/γ-AlO和NaOH/γ-AlO固体碱催化剂使3-羟甲基乙烷的收率达到86.2%,并且反232323
应条件温和。
NaCO/γ-AlO固体碱催化剂的制备:采用浸渍法制备NaCO/γ-AlO固体碱催化剂。23232323首先,按一定质量比分别称取NaCO和处理好的γ-AlO载体;然后,将NaCO用去离子232323水配成溶液,并加入γ-AlO载体,加热搅拌3~4 h(80~90?);再在100?下烘干8h,冷却23
后即得NaCO/γ-AlO固体弱碱催化剂,保存于干燥器中备用。采用相同的方法,可以制2323
备以活性炭为载体NaCO/活性炭固体碱。 23
NaOH/γ-AlO固体碱催化剂的制备:同样采用浸渍法制备NaOH/γ-AlO固体碱催化2323剂。首先,按一定质量比分别称取NaOH和处理好的γ-AlO载体,然后将NaOH用去离子23
水配成溶液,并加入γ-AlO载体,加热搅拌3~4 h(80~90?),然后在100?下烘干8h,放23
入马弗炉内在400?下焙烧3 h,冷却后即得NaOH/γ-AlO固体强碱催化剂,保存于干燥器23
中备用。采用相同的方法,可以制备以活性炭为载体NaOH/活性炭固体强碱。
3.4 金属氧化物负载KF催化合成碳酸二甲酯(DMC)
金属氧化物负载KF是甲醇与碳酸丙烯酯进行酯交换合成DMC的有效催化剂,其中,ZnO-KF固体碱催化剂的活性最高。金属氧化物负载KF固体碱催化剂的制备方法:金属氧化物先用KF水溶液浸渍后,再在383 K下干燥4 h,873K下焙烧3 h即得。
3.5 纳米超强碱KF/AlO催化合成肉桂醛 23
肉桂醛的工业传统制法是用苯甲醛和乙醛在稀碱的作用下发生Claisen-Schmidt反应缩合而成,一般以氢氧化钠作为催化剂,反应5h,产品收率最高可达到56.67%。而用纳米超强碱KF/AlO催化合成肉桂醛的反应时间短,产品收率高,该反应在30?下反应半小时,23
产率便可达到93.6%。
纳米超强碱KF/AlO催化剂的制法:采用浸渍法制备。先将30 g纳米γ-AlO溶于200 mL2323乙醇中,再加入17.4 gKF和3 g表面活性剂(按摩尔比PEG-6000:PEG-400=1:1),在65?搅拌反应3 h,然后在旋转蒸发仪中脱去溶剂,最后将所得的固体粉末在120?下干燥6 h,制得纳米超强碱KF/AlO。 23
纳米超强碱KF/AlO催化合成肉桂醛的工艺先进、方法简单、反应温度低、时间短、后23
处理方便、产品收率高;催化剂活性好,可以重复使用,另外,对环境友好,符合绿色化学的发展要求。
3.6 ZrO负载含氧酸钾盐固体超强碱 2
KNO等钾盐在ZrO上分散并经高温分解后所形成的碱的最高碱强度(H-)可达26.5,属32
于固体超强碱,在273K下的顺丁烯异构化反应中具有高催化活性。
ZrO负载含氧酸钾盐固体超强碱的制备:将钾盐(如KNO、 KCO、KHCO)和载体23233
2ZrO(Toray Ltd,Sg=120m/g)以一定比例研磨混合,按1ml/g加入蒸馏水后混捏,经383K干燥,2
再制成20~40目的颗粒。
3.7 Na-KOH/AlO固体超强碱催化剂 23
Na-KOH/AlO固体超强碱催化剂的最高碱强度H-可达43以上,碱强度H-?35的总碱23
量高达9.00 mmol/(g催化剂)。在乙稀基降冰片烯(VNB)异构化为乙叉基降冰片烯(ENB)的反应中,转化率和选择性均接近100%,催化活性极高。
Na-KOH/AlO固体超强碱催化剂的制备方法:先将100 g水合AlO干胶加到250 mL干2323
燥四口烧瓶中,然后打开氮气通路并开动搅拌,缓慢加热升温至450~500?,保持3 h后降温至365~370?;再加入20~35 gKOH,在氮气保护下搅拌反应3 h,然后在同一温度下分批加入2~3.5 g金属钠,继续反应1 h后停止。
Na-KOH/AlO固体超强碱催化剂的催化选择性极高,并且反应速度快,反应条件温和。 23
3.8 固体超强碱氧化钙催化制备生物柴油
生物柴油性质与矿物柴油非常接近,被公认为是一种可再生的清洁燃料。生物柴油以
2-植物油或动物脂肪为主要原料,经过酯交换反应得到脂肪酸甲酯类物质。CO处理过的CaO3是固体超强碱,它对酯交换反应具有较高的催化活性,并且反应速度快、反应条件温和。
氧化钙固体超强碱催化剂的制备:称取12 g碳酸铵溶于100 ml去离子水中,加入12 g氧化钙,搅拌30 min,抽滤,收集固体,在110?下烘干,研磨至60目后放入坩埚中。以15?/min的速率升温至一定温度,焙烧1.5 h。冷却至250?,取出,催化剂置于干燥器中备用。
4 小结
目前,固体超强碱催化剂的研究多处于理论研究,尚处于开发阶段,在工业上的应用很少。但固体超强碱催化剂具有超强的催化性能、易与产物分离、可循环利用、对环境友好,因此其将具有广阔的前景。
超强碱试剂种类繁多,制备方法简便,它们不但有很高的碱强度,而且还使许多反应的条件变得温和、保护设备、降低环境污染等。目前,超强碱试剂在各类有机合成中呈现出的特异的反应活性和选择性越来越引起人们的重视,正在逐渐成为大家研究的热点。
范文三:有机锌催化剂
有机锌催化剂
一、有机锌催化剂简介
锌,在化学元素周期表中位于第4周期、第IIB 族,是一种浅灰色的过渡金属,是第四" 常见" 的金属,仅次于铁、铝及铜,在现代工业中对于电池制造上有不可磨灭的地位,为一相当重要的金属。另外,锌是人体必需的微量元素之一,在人体生长发育、生殖遗传、免疫、内分泌等重要生理过程中起着极其重要的作用。
锌在自然界中的应用有很多种,世界上锌的全部消费中大约有一半用于镀锌,约10%用于黄铜和青铜,不到10%用于锌基合金,约13%用于制造干电池,以锌饼、锌板形式出现,仅约7.5%用于化学制品,主要以有机和无机化合物的形式出现,这里我们主要介绍一下有机锌化合物。
有机锌化合物在工业应用中主要有以下用途:在涂料中作助催干剂、亦称实干催干剂,用于涂料、油墨的催干、扩散剂等。在涂料中能促进漆膜底层干燥而得到坚韧而硬的涂膜,并能提高漆膜的附着力及耐候性;
另外,有机锌化合物可用作聚氨酯预聚体催化剂;
有机锌化合物具有抗腐蚀作同,在润滑油和润滑脂中使用,有防老化及抗腐性,也可用作木材防腐剂、杀虫剂等。
二、有机锌催化剂目录
异辛酸锌用作清漆催干剂、木材防腐剂、织物防水剂、杀虫剂、杀菌剂等,也在聚氨酯塑胶跑道、防水涂料及合成橡胶中作催干剂,用于替代环烷酸锌;
新癸酸锌用作清漆催干剂、木材防腐剂、织物防水剂、杀虫剂、杀菌剂等,也在聚氨酯塑胶跑道、防水涂料及合成橡胶中作催干剂,用于替代环烷酸锌;
环烷酸锌用作清漆催干剂、木材防腐剂、织物防水剂、杀虫剂、杀菌剂等,也在聚氨酯塑胶跑道、防水涂料及合成橡胶中作催干剂;
碳酸锌
乙酸锌
乳酸锌用作轻型收敛剂,配制炉甘石,皮肤保护剂,乳胶制品原料;用作制锌盐、媒染剂、木材防腐剂、试剂等;用作食品锌强化剂。
范文四:有机铋催化剂
美国领先化学品公司生产用于PU树脂浆料、硬泡、软泡、涂料、胶粘剂、密封胶和弹性体方面的安全环保型聚氨酯催化剂,已有超过30年的历史,注册商标为BiCAT,并已供应欧美市场多年。这些环保型的有机铋、有机锌和有机锆BiCAT?催化剂可在单组分和双组分体系中进行室温固化或加热固化,是替代有毒铅汞锡催化剂的极佳选择,目前已在国内广泛使用。
有机铋催化剂
产品
金属含量
BiCAT 8118
16% Bi
BiCAT 8108
20% Bi
BiCAT 8124
28% Bi
BiCAT 8106
20% Bi
BiCAT H
28% Bi
BiCAT 8
8%Bi + 8%Zn
选择性的聚氨酯凝胶催化剂;
与锡催化剂相比,对NCO基团选择性更好,制成的终产品
力学性能更佳;
诱导时间短,粘度逐渐增加。
有机锌催化剂
产品
金属含量
BiCAT Z
19% Zn
BiCAT 3228
23% Zn
与铋和锡催化剂相比,反应速度慢;
非常有效的交联催化剂,制成的终产品表面不粘手。
有机锆催化剂
产品
金属含量
BiCAT 4130
12% Zr
与锡催化剂相比,对NCO/OH反应具有高选择性;
锆催化剂与锡催化剂相比,釜中可操作时间长,产气泡少,终产品针孔少,表面光泽度高。
BiCAT和锡催化剂在胶粘剂配方中的固化特性曲线对照
BiCAT 8118:固化曲线与锡催化剂类似
BiCAT 8124:诱导期长,后期固化快速
BiCAT有机铋聚氨酯催化剂的优点
1.?????安全环保型催化剂,可取代铅汞催化剂和正在被立法废除的锡催化剂;
2.?????铋催化剂比锡催化剂有更好的耐水解稳定性,降低与水反应的选择性,在水系PU分散液中,减少水与NCO基的副反应;
3.?????促进NCO/OH反应,避免NCO副反应,减少CO2的生成;
4.?????在单组份体系中,把被水屏蔽的胺解放出来而不是促进NCO与水的反应;
5.?????铋锌组合使用有协同效果,使得配方和生产更灵活,降低能耗,提高利润;
6.?????可单独使用,或作为辅助催化剂与胺或其它有机金属化合物配合使用;
使用铋锌复合催化剂,替代汞触媒,加锌加强后固化
为达到对凝胶性态进行调节或某些客户替代汞触媒的要求,我们开发了双金属的铋锌复合催化剂BiCAT8(8%铋,8%锌)。在该种催化剂中,铋提供固化所需的凝胶速度,高选择性和高速率地生成聚氨酯,锌作为较慢的凝胶催化剂和较好的交联催化剂,可降低体系的酸度,加速反应后段,促进交联。
用户可通过改变体系中这两种金属的浓度来调节凝胶性态。如通过添加BiCAT Z (19%锌)或BiCAT3228(22%锌)增加体系中的锌,延长诱导期的釜中操作时间,促进后固化。
由于用户各自的反应体系不同,最佳铋锌比也不尽相同。保持铋量不变,通过添加不同量的锌进行试验,控制铋锌比例在1:1~1:10之间,找出最适合自己体系的铋锌比。
BiCAT聚氨酯催化剂可用于以下产品中
BiCAT催化剂是替代有毒铅汞锡催化剂的极佳选择。有机铋催化剂对催化剂体系的酸值非常敏感,只有在酸性条件下,可很好地发挥作用,当体系pH发生变化时,会对其催化活性有影响,有时变得比铅汞锡催化剂的活性低。
使用添加量建议:
1.PU树脂浆料:推荐使用BiCAT 8118,BiCAT 8108,每百份多元醇中,加入0.01-0.5%的金属铋;
2.聚氨酯涂料:推荐使用BiCAT Z,BiCAT 3228,BiCAT8或BiCAT8118,每百份多元醇中,加入1-2份催化剂;
3.聚氨酯胶粘剂:推荐使用铋锌合金的BiCAT8或更高铋含量的催化剂,每百份多元醇中,加入1-2份催化剂;
4.聚氨酯密封胶:推荐使用BiCAT H,BiCAT8或BiCAT 8118,每百份多元醇中,加入1-2份催化剂;
5.聚氨酯弹性体:推荐使用BiCAT3228,或配合使用BiCAT 8和BiCAT Z,其中Zn的量可控制为Bi的1-10倍,这取决于客户对反应体系粘度和可操作时间的具体要求,或使用更高铋含量的催化剂,每百份多元醇中,添加0.01-0.5%的金属铋;
6.聚氨酯软泡:推荐使用BiCAT 8106。
范文五:有机金属催化剂
前 言
22现代有机合成中过渡金属催化偶联反应是形成碳(sp)?碳和碳(sp)?杂键的重要反应类型,近年来,因为其简洁、高效而经济,己经成为合成化学中不可缺少的有效途径,在药物分子、除草剂、天然产物、液晶材料、染料、聚合物等各
[1-3]种有机物合成方面均有着广泛的应用,并为社会带来了显著的经济效益。
目前过渡金属催化的C?C形成反应主要包括Kumuda、Negishi、Stille、Sonogashira、Suzuki和Heck偶联反应等 (Fig.1)。
图1 交叉偶联反应
Fig. 1 Cross-coupling reaction
在过渡金属催化的芳基偶联反应中,由日本北海道大学的Suzuki、Miyaura等人于1981年提出的在钯(0)催化下,有机硼酸作为亲核基团,与卤代烃或假卤代烃(如:含三氟磺酸基的化合物)等碳有机亲电试剂在碱存在下进行的C?C偶联
[4]反应被称为Suzuki-Miyaura偶联反应,是合成C?C键的有效方法之一。反应通式见Fig. 2。
图2 Suzuki-Miyaura偶联反应
Fig. 2 Suzuki-Miyaura coupling reactions
该反应因为具有反应条件温和,区域选择性和立体选择性好,副反应少并且反应的反应物和副产物一般是无毒或低毒的无机硼酸,对环境友好,副产物容易从目标产物中分离出来,反应操作简单安全;其次相对于其它的有机金属试剂而言,有机硼试剂对醛基、酯基、氰基、硝基、羟基等在内的多种活性官能团兼容性好,受空间位阻影响不大、产率高以及芳基硼酸原料来源广泛且容易制备,而且对空气、水不敏感等优点而得到普遍应用的Aryl-Aryl键偶联方法,是现代有
[5]机合成最重要的手段之一,被应用到很多有机分子的合成领域。目前,对该反应的研究主要集中在反应底物的拓展、寻找新的催化体系、减少或不使用复杂昂贵的配体,优化反应体系、使用新的实验技术如微波、无溶剂操作等方面。
22另外,过渡金属催化的碳(SP)?碳和碳(SP)?杂催化偶联反应,通常都需要使用各种结构复杂的配体来提高催化剂的催化活性,过去二十年来,富电子的含膦(磷)配体、卡宾配体、含氮配体以及其它含杂原子配体的使用得到了较快发展,使得 Suzuki-Miyaura偶联反应也取得了重大突破。但是,钯催化剂价格昂贵,镍有毒性,并且对于许多配体或催化剂体系而言,存在着结构复杂、制备条件苛刻、容易失活及反应后难以回收使用的问题,这就使其应用于大规模反应受到了很大的限制。因此,开发价廉易制备的配体或催化剂体系,使其能够具有温和的催化反应条件、足够高的反应活性,降低催化剂的用量,扩展偶联反应应用范围,也是目前催化偶联反应中迫切需要解决的问题。
1.2 钯催化Suzuki-Miyaura偶联反应研究进展
1.2.1 Suzuki-Miyaura偶联反应机理
对 Suzuki-Miyaura偶联反应目前公认的机理如 Fig.1.5 所示。在一个催化循环过程中,通常涉及到氧化加成?转金属化?还原消除这一系列步骤,尽管每一步都涉及到其它一些具体的过程,例如配体交换等等,但这些步骤的中间体已经被证明确实存在。其它由 Ni(0)、Fe(I)等催化的交叉偶联反应也通常具有类似的
[23]催化循环。
图1.5 钯催化的Suzuki-Miyaura 偶联反应机理
Fig. 1.5 The machnism of palladium-catalyzed Suzuki-Miyaura coupling reaction
1.2.2 含膦配体钯催化体系
Pd(PPh)是最早应用于Suzuki-Miyaura偶联反应的膦配体催化剂,其热稳34
定性好,在反应中不易分解,其它的配体有n-BuP、(MeO)P、PCy以及双齿膦333配体PhP(CH)PPh(dppe)、Ph(CH)PPh(dppp)、Ph(CH)PPh(dppb),222222322234л-(CH)PhPFe(dppf)等。但由于这些配体对水比较敏感,副产物多,反应操作5422
需在氮气保护下进行并且催化剂用量较高(1?10 mol%),而且难以回收,若应用于大规模反应则成本太高。
1998年Fu等发现Pd(dba)/P(t-Bu)做催化剂,较广范围(包括给电子的)的233
氯代芳烃与缺电子、电中性和给电子的芳基硼酸在80 ?С发生Suzuki反应,均可得到良好的产率(Fig.1.6)。 但是,在实际应用过程中三烷基膦配体对空气十分敏感,很容易被氧化而失去配位能力,钯催化剂就会失去相应的高催化活性。所以,有机膦/钯配合物催化的偶联反应一般都需要在惰性气体保护下进行,从而给实际操作带来很大困难。为了解决这一问题,Fu等又提出简单而有效的策略:通过磷的质子化作用将对空气敏感三烷基膦转化成的在空气中可以稳定存在的磷盐。实践证明在Suzuki-Miyaura偶联反应中对空气稳定的[(t-Bu)PH]BF是对空34
[24]气敏感三烷基膦的有效替代物。
图1.6 三(二苯基亚甲基丙酮)合二钯/三叔丁基膦催化的Suzuki-Miyaura偶联反应
Fig. 1.6 Pd(dba)/P(t-Bu) catalyzed Suzuki-Miyaura coupling reaction 233
Buchwald课题组对于联苯结构的二烷基单膦配体2?11(Fig.1.7)用于Suzuki-Miyaura 偶联反应,进行了全面的报道,发现非常低的催化剂负载量能有效的催化含氮、氧和硫的杂芳基卤代物或大位阻芳卤与含氮、氧和硫杂环芳基硼
[25]酸或乙烯基硼酸的偶联反应。
图1.7 大位阻的联苯型膦配体
Fig. 1.7 Hindered biarylphosphine ligands
2000年,Beller等报道了原位钯催化体系,在体系中采用膦配体,用
5Pd(OAc)/P(OR)能高效地催化不活泼的溴代芳烃的偶联 (TON:8.2 , 10),同23
时实现了氯代芳烃进行偶联反应。实验发现:偶联反应成功的关键因素是合适的
[26]碱性条件(如:CsF)及Pd/P的配比。
图1.8 1,6,二烯Pd(0)单膦配合物
Fig. 1.8 1,6-diene Pd(0) single-phosphine complexes
后来该组以烯丙基醚、烯丙基胺、四甲基二烯基二硅氧烷分别与PdPCy反3应生成1,6,二烯Pd(0)单膦配合物12?16 (Fig.1.8),催化氯代芳烃的Suzuki-Miyaura 偶联取得了好的结果。例如在0.05 mol%的16催化下,100?С时在THF中,KF和KPO存在下2?氰基氯苯、氯苯、4?甲氧基氯苯与苯基硼酸34
[27]反应22 h,产率分别达到97%、87%和72%。
Santelli等人报道了一种新型四齿磷配体Tedicyp(Fig.1.9),能高效地催化活化氯代芳烃的Suzuki-Miyaura 偶联反应。作者认为四个二苯基磷基位于环戊烷
[28]的同一面,与钯配位紧密,增加了催化剂体系的稳定性。
图1.9 四齿磷配体
Fig. 1.9 Tedicyp ligand
2004年,Colacot等报道了空气中稳定、高效的催化剂CpFe(PR)PdCl (R 2222= i-Pr,t-Bu)18应用于富电子和大位阻的氯代芳烃的Suzuki-Miyaura 偶联反应
[29](Fig.l.10)。
图1.10 二茂铁双膦的氯化钯配合物催化Suzuki-Miyaura 偶联反应
Fig. 1.10 CpFe(PR)PdCl catalyzed Suzuki-Miyaura coupling reaction 2222
Verkade等制备了一系列新型大空间位阻的双环笼状配体19a?19f,筛选出具有高催化活性的配体19d。Pd(OAc)/19d体系能有效地催化溴代芳烃、氯代芳2
[30]烃与苯硼酸的Suzuki-Miyaura偶联反应,取得分离产率大于90%。
图1.11 双环三胺膦配体/ Pd(OAc)催化Suzuki-Miyaura 偶联反应 2
Fig. 1.11 Bicyclic triaminophosphine ligands/Pd(OAc) catalyzed Suzuki-Miyaura coupling 2
reaction
虽然含膦的Pd催化剂体系在Suzuki-Miyaura偶联反应中取得了很大的进展,甚至通常反应很困难的富电子氯代芳烃为底物时也能被成功地催化。但是,由于有机膦配体对空气十分敏感,很容易被氧化而失去配位能力,有机膦?钯催化体系就会失去相应的高催化活性。所以,有机膦钯配合物催化的偶联反应一般都需要在惰性气体保护下进行,从而会很大程度上限制该类催化剂的广泛应用。因此,发展无膦的对空气和水都不敏感的催化剂具有十分重要的研究意义和应用前景[31-33]。
1.2.3 N-杂环卡宾钯催化体系
具有亲核性的N?杂环卡宾(N-NHC)配体被认为是具有两电子给予体而又几乎没有反馈π?键的、中性的优良有机配体由于其较高的富电子性及良好的稳定
[34]性及配位性能,这一特性与给电子的膦配体相似,可称之为“仿膦配体”。这类配体与钯配位牢固,Pd-NHC键的稳定性降低了过量的配体的使用,而且这类配体的富电性特性加快了氧化加成基元反应的速率,其立体位阻又有利于形成单卡宾物种,提高了还原消除基元反应的速率。与膦配体相对而言,NHC具有合成工艺简单,稳定性高等特点。因此,在 Suzuki-Miyaura偶联反应中的应用也取得了一定的进展。但是从实用的角度看,它同样存在着化学性质不稳定,成本高等缺点。
图1.12 N?杂环卡宾配体
Fig. 1.12 N-NHC ligands
1998年,Herrmann等发现化合物20在甲苯/120?С/KCO条件下,可催化23
[35]溴代芳烃和缺电子的氯代芳烃的Suzuki-Miyaura 偶联反应。
Nolan等报道了在1.5 mol% Pd(dba)/1.5 mol%卡宾21 dioxane/CsCO/80 ?С2323条件下,4?氯甲苯与苯基硼酸的偶联反应,可得到59%分离产率。考虑到卡宾
[36]21遇空气和水都不稳定,他们又制备了咪唑盐22,原位生成卡宾,与不同的钯源一起组成催化体系,Pd(OAc)/22a,Pd(dba)/22b的催化体系能催化带各种223
[37]取代基和有位阻的氯代芳烃与芳基三氟甲基磺酸酯的偶联反应。
Altenhoff等发展了一种催化体系,在催化量叔丁醇钠存在下,用KH/THF处理咪唑盐23原位生成N-杂环卡宾配体,以Pd(OAc)为钯源,室温下可高效2
[38]地催化氯代芳烃与芳基硼酸的偶联,生成具有较大位阻的联苯类化合物。
Ozdemir等制备了含饱和咪唑环的溴化二烷基咪唑盐LHX (24 a-d)。在反应中原位生成卡宾,和Pd(OAc)催化氯代芳烃与苯基硼酸的偶联,反应无诱导期。2
以1.5 mol% LHX配体24d/1.5 mol% Pd(OAc)为催化剂,KCO/dioxane/60 ?С223
[39]条件下,4?甲氧基氯苯与苯基硼酸反应2h,可以95%的产率得到偶联产物。
2002年Herrmann等人通过配体交换反应制备出了金刚烷基卡宾钯配合物25和26,当配合物25用于催化4?氯甲苯与苯硼酸的偶联反应时,仅仅获得20%的转化率;而配合物26在室温条件下可以高效地催化各种氯代芳烃与芳基硼酸
[40]的Suzuki-Miyaura偶联反应,产率在75?99%。
图1.13 N-金刚烷基卡宾钯配合物催化氯代芳烃Suzuki-Miyaura偶联反应 Fig. 1.13 N-Adamantyl NHC-Pd complex catalyzed aryl chlorides Suzuki-Miyaura coupling
reactions
2003年Nolan等人利用带有六元环状卡宾钯配合物27a 和27b催化氯代芳烃与芳基硼酸的Suzuki-Miyaura偶联反应。配合物27a在室温条件下用异丙醇做溶剂在较短的反应时间内 (50?120 min),可以有效地催化各种氯代芳烃(其中包括邻位有一定空间位阻的氯代芳烃)与芳基硼酸的Suzuki-Miyaura偶联反应,产
[41]率在77?95%。
图1.14 六元环状卡宾钯配合物催化氯代芳烃Suzuki-Miyaura偶联反应 Fig. 1.14 Six-cyclic carbene palladium complex catalyzed Suzuki-Miyaura coupling reactions
1.2.4 环钯化合物催化体系
环钯化合物是一类分子内通过一个或两个中性给电子原子所稳定的含有Pd-C键的环金属化合物。其形成条件是分子内部存在能与金属配位的基团,通过配位使相邻碳键活化,实现碳与金属键合,形成一类具有C-M σ键的四元、
[42]。 五元、六元或更大环螯合物
环钯化合物由于具有容易制备、对热和空气稳定、活性较高、寿命较长等优点,被认为是适合循环使用的催化剂。在这一领域的最初应用是由Beller、Herrmann等于1995年发现热稳定的环钯化合物28在催化苯基硼酸与4?乙酰基氯苯、4?甲氧基溴苯、4?乙酰基溴苯的偶联反应时有较好的催化效果,TON值
[43]分别达到2100、7600和74000。
后来Bedford、Hazelwood等人合成了几种钳形化合物29和P、O环钯二聚体30a-d,在催化带给电子取代基的4?甲氧基溴苯与苯基硼酸的偶联反应时,都
456表现出较好的催化效果,催化剂30a-d的TON分别为3×10,2.9 ×10,2.6 ×10,
6[44,45]4.2×10。不久Cole-Hamilton等人用Pd与膦配体形成的环钯化合物31在催
[46]化Suzuki-Miyaura偶联反应中也表现出了很好的催化活性。
当然,具有较好催化活性不仅仅局限于P?供电配体邻位环钯配合物,最近一些化学家研究发现N?供电配体环钯配合物32a也可用于催化溴代芳烃代物的
偶联反应。值得一提的是,Milstein等合成了在空气中稳定存在的亚胺环钯二聚体,在空气中应用33a催化给电子的溴代芳烃和苯基硼酸的偶联反应可以得到高
5[49][47,48]于10的TON值。亚胺是有效的有机配体。
同时Najera等人也合成了一系列对空气和水稳定的亲水性环钯肟衍生物34a-c,在有机相和水相中都能有效催化溴代芳烃或氯代芳烃与苯基硼酸的偶联。其中配合物34b在水相中能够催化氯代芳烃的Suzuki-Miyaura偶联反应,并且在
[50,51]添加TBAB后,TON值达到9000,而TOF值达到3850。
Dupont和Monteiro等人发现在空气中稳定存在的无膦型含硫环钯化物35能够较好地催化4?甲基溴苯和苯基硼酸的偶联反应。并且在室温下催化溴代芳烃
[52]及活化氯代芳烃的Suzuki-Miyaura偶联反应时,分离产率大于90%。
图1.15 环钯配合物
Fig. 1.15 Cyclo-palladium complexes
Bedford等合成了含胺及亚胺的环钯化二聚体及其膦加成物36和37,发现
[53]在催化氯代芳烃和苯基硼酸的偶联反应中表现出非常好的反应活性。最近,该课题组报道二聚体30和38与三环己基膦形成相似的加成物这是目前所知文献
[54,55]报道的最好的催化氯代芳烃的偶联反应的催化剂之一。Liu等合成了含六元螯合环的环钯化二聚体39及其膦加成物40,发现以KCO为碱,在回流的乙醇23
6[56]中,39a和39b催化4?甲基溴苯和苯基硼酸的偶联反应TON值可达10。
Wu等人于1994年首次报道了二茂铁亚胺环钯类化合物41的合成,并于2001年将其应用于溴代芳烃中的Suzuki-Miyaura 偶联反应,获得了较好的产率[57,58]。
图1.16 亚胺环钯配合物
Fig. 1.16 Imine cyclo-palladium complexes
在研究含氮配体与钯盐形成的络合物对Suzuki-Miyaura偶联反应的催化活性中,亚胺(Schiff base)类配体得到较多关注,Carcia-Ruano等报道了配体42与醋酸钯形成endo型(C?N在环内)含C-Pd ,键五员环配合物43 (Fig.1.17),将其
[59]应用到Suzuki-Miyaura偶联反应,取得了很好的效果。
反应式 1.17 亚胺环钯配合物的合成
Fig. 1.17 Preparation of imine cyclo-palladium complexes
[60]环钯化合物根据金属核的数目分为单核环钯化合物和双核环钯化合物。在没有其它配体存在时一般生成双核环钯化合物,而在有其它配体存在的条件下则解离生成单核环钯化合物。单、双亚胺配体与钯形成的环钯二聚体配合物,在常见的有机溶剂中通常难以溶解,通常加入其它配体将其解聚为单体以便于常规波谱鉴定或用于其它有机合成。用于解聚的配体有:胺、吡啶、吡唑、三苯膦、乙酸丙酮等。
图1.18 双核和单核环钯配合物
Fig. 1.18 Dinuclear and mononuclear cyclopalladat
具有多配位中心的亚胺配体,其配位中心可处于一个或多个芳环上。1996 年
[61]Chakladar、Vila等利用对苯双亚胺合成了具有对称结构的环钯配合物44。
图 1.19 对称双亚胺环钯配合物
Fig. 1.19 Symmetric diimine cyclopalladated complex
Cheprakov等研究发现,亚胺环钯化合物在Heck及Suzuki-Miyaura偶联反应中都有良好的催化活性。作者认为亚胺环钯化合物中配体只是作为钯的载体,和活性并无太大的关系,因此没有必要去设计复杂的配体,而廉价的含N化合
[62]物(席夫碱、肟、苄胺等)是最好的选择。
1.2.5 含氮配体钯催化体系
膦配体/钯催化体系的的确具有很高的反应活性,但膦配体自身存在合成困难、具有毒性,价格相对昂贵,对空气和水敏感,严格要求无水无氧实验操作等问题,并且在反应过程中P?C键在高温时会发生断裂,容易生成副产物,给产物的分离带来麻烦。这些问题的存在不仅给实际操作带来很大困难同时对环境造成污染。近年来,为了克服膦配体的缺陷,一些非膦配体的使用在一定程度上克
[63]服了膦配体的不足,其中含氮配体是比较重要的一种。因此,关于筛选在空气和水中可以稳定存在、反应条件温和,并且经济高效的配体或高效环保的催化体系的研究引起了科研工作者的重视,这几年取得了很多突破。一些氮配体(如N?杂环卡宾)是非膦配体中重要成员,引起了人们的重视,实践证明含氮配体同样可以有效地稳定活化过渡金属催化剂。
(1) 噁唑类配体
2002年,Boykin等人合成了一系列含有唑啉环的单齿配体45a-f以及双齿配体46和47,用Pd(OAc)作为钯源,在80 oC下原位催化Suzuki-Miyaura偶联2
反应,结果发现配体的空间结构以及配体上的取代基的电子效应对偶联反应有着明显的影响。当配体45e和45f苯环上的取代基团R为吸电子基时(如:-CN和-NO)2催化活性明显好于苯环上带有供电子基团的配体45a-d。而对于双齿配体46和47来说,用47作为配体时,催化剂的催化活性要好于唑啉环上N-邻位有一定空
间位阻的配体46。而配体47与配体45e和45f相比较,催化溴代芳烃的偶联反应中效果也是最好的,产率在75-99%之间。但是需要2 mol%的Pd(OAc),催化2
[64]剂用量较高。
图1.20 噁唑类配体/Pd(OAc)催化的Suzuki-Miyaura偶联反应 2
Fig. 1.20 Oxazole ligands/ Pd(OAc) catalyzed Suzuki-Miyaura coupling reactions 2
Gade等设计了一种新型钳状二噁唑吡咯配体48。该配体在与PdC1(COD)反2应后生成了三个氮原子配位的配合物49。由于三个氮原子与钯配位,钯原子具有较高的电子云密度,氧化加成反应速率大幅度提高。该配合物催化4?乙酰基溴苯和4?甲氧基溴苯与苯硼酸的Suzuki-Miyaura 偶联反应TON值分别达到82000和54000。分别在70 oC 或110 oC的温度下催化其它溴代芳烃与苯硼酸的偶联反应
[65]时,产率在54?97%之间。
图1.21 二噁唑吡咯钯配合物催化的Suzuki-Miyaura偶联反应 Fig. 1.21 Suzuki-Miyaura coupling reactions catalyzed by bis(oxazolinyl)pyrrole]palladium
complex
2006年,Lee合成了含有二茂铁结构单元的二噁唑双齿配体,与PdCl配位2后形成配合物50 (顺式) 和51 (反式) 两种。但是它们用于催化Suzuki-Miyaura偶联反应的效果没有明显差别。其中配合物50能够有效地催化溴代芳烃的偶联反应,而对于催化活化的氯代芳烃4?氯苯乙酮与苯硼酸的偶联反应,在80 oC下仅获得26%的产率,即使反应温度提高到110 oC,反应产率也没有明显提高,仅
[66]为45%的产率。
图1.22 配合物50催化Suzuki-Miyaura 偶联反应
Fig. 1.22 Suzuki-Miyaura coupling reactions catalyzed by complex 50
(2) 胺类配体
2003年Boykin利用二环己基胺(CyNH)与醋酸钯制得2
trans-Pd(OAc)(CyNH)。这个催化剂,在室温下可以实现带吸电子基团溴苯的222
Suzuki-Miyaura偶联反应。提高反应温度至80 oC时,可以实现带供电子基团溴
[67]苯的Suzuki-Miyaura偶联反应,例如4?甲氧基溴苯就可以有94% 的产率。
图1.23 二环己基胺的醋酸钯配合物催化Suzuki-Miyaura偶联反应
Fig. 1.23 Suzuki-Miyaura coupling reactions catalyzed by trans-Pd(OAc)(CyNH) 222
三乙烯二胺53 (DABCO)也是含氮的简单二胺化合物,Li等把该化合物与醋酸钯组成催化剂体系应用于碘代芳烃、溴代芳烃和一些活性较高的氯代芳烃的Suzuki-Miyaura 偶联反应,结果发现该催化体系具有较高的催化能力。在丙酮中,
110 oC下催化碘苯与4?氯苯硼酸的Suzuki-Miyaura 偶联的TON值达到了
[68][69]950000。当以PEG400或PEG-400/水(V : V = 1 : 5) 做溶剂,在加入添加剂TBAB后,可以实现氯苯的偶联反应产率为70%。
图1.24 三乙烯二胺/Pd(OAc) 催化Suzuki-Miyaura 偶联反应 2
Fig. 1.24 Suzuki-Miyaura coupling reactions catalyzed by DABCO/Pd(OAc) 2
(3) 席夫碱配体
2001年Nolan课题组报道了将一系列的DAB?R(二氮杂丁二烯)类化合物54?60作为配体与Pd(OAc)(3 mol%)作为有效的原位催化体系用于催化卤代芳烃2
与苯硼酸的偶联反应。研究发现配体上取代基团的电子效应对催化Suzuki-Miyaura偶联反应有明显的影响,当配体上R基团为供电子性的烷基时,催化活性大大提高,所得的产率也得到相应的增加。其中环己基二亚胺与二醋酸钯组成的催化体系催化活性最高,用于各种溴代芳基的Suzuki-Miyaura偶联反应时可获得90?99%的分离产率。催化缺电子的氯代芳烃时也能得到较好的产率,而对于富电子的氯代芳烃结果较差,这可能与烷基强的给电子能力有关。当R基为金刚烷55作配体时产率可达到93%,R为芳基时效果稍差。这是一种效果
[70]很好的新型非膦配体。但是反应过程中Pd(OAc)的用量较高 (3 mol%)。 2
图1.25 席夫碱配体/Pd(OAc)催化Suzuki-Miyaura偶联反应 2
Fig. 1.25 Suzuki-Miyaura coupling reactions catalyzed by Schiff base ligands/ Pd(OAc) 2
(4) 吡啶类配体
2003年,Nájera等人报道了二?(2?吡啶)甲基胺衍生的配体与二氯化钯络合形成的61,在CHOH/HO的混合溶剂中,KOH为碱,用1?0.00076 mol%催化32
剂催化溴苯、碘苯与苯基硼酸的Suzuki-Miyaura偶联反应,获得了较好的产率37?99%。如果在上述反应体系中加入添加剂TBAB,那么对于氯苯与苯硼酸的
[71]偶联反应也可以进行。
图1.26 配合物61催化Suzuki-Miyaura偶联反应
Fig. 1.26 Suzuki-Miyaura coupling reactions catalyzed by complex 61
2008年,Thore等人利用2?(2?吡啶?3?乙胺基)乙基磺酸钠62为配体,与二氯化钯作用,成功实现了室温下水相中的Suzuki-Miyaura偶联反应。但是,仅仅是对碘代和溴代芳烃具有较好的催化效果,产率在84?90%之间。并且在实验中发现,如果在反应中加入配体的量与碘代或溴代芳烃量是等当量时,反应体系中
可以不需要加入额外的碱,偶联反应就可以顺利的进行,在这里吡啶基团可能即
[72]起到配位作用,同时也可以作为碱使用。
图1.27 配合物62催化的Suzuki-Miyaura偶联反应
Fig. 1.27 Suzuki-Miyaura coupling reactions catalyzed by complex 62
近来,Xi等人合成喹喔啉联吡啶,利用吡啶上的N原子和喹喔啉上的N原子作为双齿配体与PdCl配位形成配合物63a-c,并用于催化Suzuki-Miyaura偶2
联反应,结果发现配体中吡啶环上取代基的电子效应对催化活性有着明显的影响。一般说来,带有吸电子基团的配体活性较差(63c < 63b="">< 63a)。其中配合物63a="" 利用0.1="" mol%="" 的催化剂量,在甲苯溶液中80="">
[73]烃与苯硼酸的偶联反应,产率在96?99%之间。
图1.28 配合物63a催化Suzuki-Miyaura偶联反应
Fig. 1.28 Suzuki-Miyaura coupling reactions catalyzed by complex 63a
(5) 腙类配体
Fujita等制备了具有腙结构的一类化合物64和65,并把它们与醋酸钯配位组成催化体系,在室温和空气气氛下实现了水相中的Suzuki-Miyaura 偶联反应,产率在24?99%之间,在升高温度的条件下,配体64和醋酸钯可以催化缺电子
[74]的氯苯,达到67%的产率。
图1.29 腙类配体/Pd(OAc)催化的Suzuki-Miyaura 偶联反应 2
Fig. 1.29 Suzuki-Miyaura coupling reactions catalyzed by hydrazone ligands/Pd(OAc) 2
(6) 胍类配体
2007年,Zhang等人利用胍作为配体66,在乙醇和水混合溶剂中,与Pd(OAc)2作用考察了催化Suzuki-Miyaura 偶联反应的活性,发现在室温条件下,上述体系能够有效地催化碘代或溴代芳烃的偶联反应,但是对于氯代芳烃的催化活性较
[75]差,即使是升高反应温度至80 oC,产率提高并不明显。
图1.30 胍类配体/Pd(OAc)催化Suzuki-Miyaura 偶联反应 2
Fig. 1.30 Suzuki-Miyaura coupling reactions catalyzed by guanidine ligand/Pd(OAc) 2
(7) 卟啉配体
Kostas等人首次将水溶性的卟啉钯配合物67应用于Suzuki-Miyaura偶联反应中。以KCO为碱,催化剂与底物的比值为1:1000,以水做溶剂,100 oC下反应23
4 h,该催化体系能在空气条件下催化溴代芳烃代物和苯硼酸的Suzuki反应,产率
[76]为80?100%。
图1.31 基于卟啉的新型四亚胺盐钯配合物
Fig. 1.31 Novel tetraimidazolium salt based on a porphyrin
(8) 吡唑类配体
双齿配体中也有杂环吡唑上的N原子和亚胺上的N原子同时做配位原子的
例子。Sarkar合成了一系列吡唑?希夫碱配体68?71,能够很好地催化各种溴代
芳烃的Suzuki-Miyaura偶联反应,对于缺电子的氯苯及4?甲基氯苯的
Suzuki-Miyaura反应,产率达到67?95%。但是对于富电子的4?甲氧基氯苯,却
[77]没有催化活性。
图1.32 吡唑?希夫碱配体/Pd(dba)催化Suzuki-Miyaura 偶联反应 23
Fig. 1.32 Suzuki-Miyaura coupling reactions catalyzed by pyrazole-Schiff base ligands
(9) 喹啉类配体
Guo等人报道了利用8?羧基喹啉与二价钯形成的N,O?双齿螯合配合物,
在0.01 mol%催化剂量下,50 oC下,乙醇和水作为混合溶剂能够有效的催化碘
代和溴代芳烃的偶联反应。TON值可达到10000,但是这一体系无法催化氯代芳
[78]烃的偶联反应。
图1.33 N, O?双齿螯合配合物72催化Suzuki-Miyaura 偶联反应 Fig. 1.33 Suzuki-Miyaura coupling reactions catalyzed by N, O-bidentate chelate complex 72
1.2.6 其它配体钯催化体系
(1) 硫尿类配体
非磷配体中除了N配体以外,S原子也被用做配位原子来参与催化过程。但是,由于S原子与Pd的结合能力过强,常会由于过分配位而使Pd中毒,反而阻止了催化循环。尽管如此特定结构中的S原子仍可以有好的催化活性的。2004年,Yang等人报道了硫尿素73作为配体可以在异丙醇和水的混合体系中,在80 oC与PdI作用来催化碘苯和溴苯的Suzuki-Miyaura偶联反应,产率在70?99%2
[79]之间。
图1.34 硫尿类配体73/PdI催化Suzuki-Miyaura 偶联反应 2
Fig. 1.34 Suzuki-Miyaura coupling reactions catalyzed by thiourea ligand 73/PdI 2
2006年,双硫尿素74b 被成功应用于碘代以及溴代芳烃的Suzuki-Miyaura
[80]偶联反应,产率在27?99%之间,并且活化的氯代芳烃也获得了中等的产率。
图1.35 双硫尿素配体74b/PdI催化Suzuki-Miyaura 偶联反应 2
Fig. 1.35 Suzuki-Miyaura coupling reactions catalyzed by bithiourea ligand 74b/PdI 2
(2) 1,3?二羰基类配体
2007年,Guo等人利用简单的二羰基化合物作为配体,发现只有结构最简单的乙酰基丙酮75与0.01 mol% Pd(OAc)作用,在50 oC下催化碘代或溴代芳2
[81]烃的效果是最好的,而其它配体的活性较低。
图1.36 非膦二羰基类配体/Pd(OAc)2 催化Suzuki-Miyaura偶联反应 Fig. 1.36 Suzuki-Miyaura coupling reactions catalyzed by phosphine-free dicarboxylic ligands
1.4 本论文的研究思路
由于Suzuki-Miyaura偶联反应条件温和、官能团兼容性好、受空间位阻影响不大、产率高以及芳基硼酸经济易得、无毒且对潮气不敏感等优点,已经成为形成碳?碳键的最有效方法之一,已广泛地应用于合成天然产物、药物中间体、液晶和功能材料中。目前广泛使用膦钯配合物催化Suzuki-Miyaura偶联反应,由于膦配体本身具有一定的局限性,因此一直致力于寻找一些优良的非膦配体。氮配体本身具有的优点如:结构类型多样、对空气和水稳定、价格低廉、容易制备,因此成为关注的热点。

