


 亖呉?盀
亖呉?盀
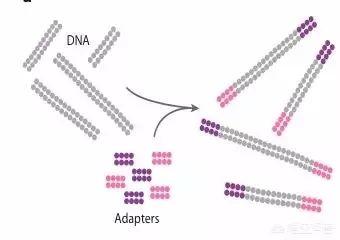
范文一:pcr 反应步骤及注意事项
实验中的一些好习惯
1 加入试剂之前,把它混匀一下,以免放置时间长了浓度不均
2 移液枪用完之后要归到最大计量的位置,防止久而久之弹簧失去弹性
3 一定要记着关水浴箱,切记切记
4 多和大家讨论,同时多关注别人讨论的经验,这几乎是最快提高的捷径了
5 所有的试剂都自己配,出了问题才好找原因
6 所有的PCR试剂都应小量分装,如有可能,PCR反应液应预先配制好,然后小量分装,-20?保存备用,以减少重复加样次数,避免污染机会。
7 PCR试剂与反应液应与样品及PCR产物分开保存,不应放于同一冰盒或同一冰箱。 试验前准备,防止RNA酶污染的措施:
1. 所有的玻璃器皿均应在使用前于180?的高温下干烤6h或更长时间。
2. DEPC处理
(1)0.1%DEPC水:100ml超纯水加入0.1ml DEPC,充分混匀,高压灭菌。
(2)Tip头(枪头)、EP管等在提RNA过程中及做RT时接触RNA的器材(包括1ml、200μl、20μl Tip
头;EP管和PCR反应管等);用0.1%的DEPC水(1000 ml超纯水加入1ml DEPC)37?浸泡过夜,高压
灭菌。
枪头盒插好枪头后,加入1-2ml的0.1%的DEPC水,再灭菌就行了。
3. 有机玻璃的电泳槽等,可先用去污剂洗涤,双蒸水冲洗,乙醇干燥,再浸泡在3% H2O2 室温10min,
然后用0.1% DEPC水冲洗,晾干。
4. 配制的溶液应尽可能的用0.1% DEPC,在37?处理12h以上。然后用高压灭菌除去残留的DEPC。不能
高压灭菌的试剂,应当用DEPC处理过的无菌双蒸水配制,然后经0.22μm滤膜过滤除菌。
5除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管及样进枪头等均应一次性使用。
6(试剂尽量分装,不要原瓶多次取用。
7. 操作人员戴一次性口罩、帽子、手套,实验过程中手套要勤换。
8. 设置RNA操作专用实验室,所有器械等应为专用。
标本处理区,包括扩增摸板的制备;
PCR扩增区,包括反应液的配制和PCR扩增;
产物分析区,凝胶电泳分析,产物拍照及重组克隆的制备。
各工作区要有一定的隔离,操作器材专用,要有一定的方向性。如:标本制备?PCR扩增?产物分析?
产物处理。
切记:产物分析区的产物及器材不要拿到其他两个工作区,PCR产物分析所用加样器不能拿到其它两个
区;
使用一次性吸头,严禁与PCR产物分析室的吸头混用,吸头不要长时间暴露于空气中,避免气溶胶
的污染。
1 引物的设计、合成
应用Permier5.0软件,参照GenBank中注册的Mass41株IBV全基因序列(GeneBank索引号:gi,57116625,gb,AY851295.l,)中N基因序列及3’端非编码区(UTR)序列,设计了一对引物NP1和NP2,其引物扩增产物包括全部N基因以及IBV基因组3’端非编码区(UTR)的基因片断,跨幅为1404bp。引物由北京赛百盛生物公司合成,序列见表:
NP1 5’CCA GGG GAA AAC TTG TGA GGA 3’
NP2 5 AAA TCT AAG GGT CTA CCG TTC GTT3’
注意:
(1)引物: 引物是PCR特异性反应的关键,PCR 产物的特异性取决于引物与模板DNA互补的程度。
理论上,只要知道任何一段模板DNA序列, 就能按其设计互补的寡核苷酸链做引物,利用PCR就可将模
板DNA在体外大量扩增。
设计引物应遵循以下原则:
?引物长度: 15-30bp,常用为20bp左右。
?引物扩增跨度: 以200-500bp为宜,特定条件下可扩增长至10kb的片段。
?引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。ATGC最
好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列。
?避免引物内部出现二级结构,避免两条引物间互补,特别是3'端的互补,否则会形成引物二聚体,产生
非特异的扩增条带。
?引物3'端的碱基,特别是最末及倒数第二个碱基,应严格要求配对,以避免因末端碱基不配对而导致PCR
失败。
?引物中有或能加上合适的酶切位点, 被扩增的靶序列最好有适宜的酶切位点, 这对酶切分析或分子克
隆很有好处。
?引物的特异性:引物应与核酸序列数据库的其它序列无明显同源性。引物量: 每条引物的浓度0.1,1umol
或10,100pmol,以最低引物量产生所需要的结果为好,引物浓度偏高会引起错配和非特异性扩增,且可
增加引物之间形成二聚体的机会。
(2)引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散
的常见原因。有些批号的引物合成质量有问题,两条引物一条浓度高,一条浓度低,造成低效率的不对称
扩增,对策为:?选定一个好的引物合成单位。?引物的浓度不仅要看OD值,更要注重引物原液做琼脂
糖凝胶电泳,一定要有引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无
条带,此时做PCR有可能失败,应和引物合成单位协商解决。如一条引物亮度高,一条亮度低,在稀释引
物时要平衡其浓度。?引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏部分,导致引物变质
降解失效。?引物设计不合理,如引物长度不够,引物之间形成二聚体等。
2 Trizol 试剂法抽取RNA
Trizol法适用于人类、动物、植物、微生物的组织或培养细菌,样品量从几十毫克至几克。用Trizol法提取的总RNA绝无蛋白和DNA污染。
关于Trizol Reagent需要的试剂
(1)(氯仿 (分析纯)
(2)(异丙醇(分析纯)
(3)(75%乙醇。要求用分析纯无水乙醇并用0.01%的DEPC处理过的无Rnase的水稀释。 (4)(无Rnase的水RNase-free water即0.1%DEPC水:100ml超纯水加入0.1ml DEPC,充分混匀,在37?过夜,高压灭菌(150? 3小时)。
(5)(一次性塑料手套
(6)(注意:DEPC有致癌之嫌
2.1 病料的处理:
无菌取病鸡的肾脏、肺和气管,按1:2的比例加入生理盐水将组织充分研磨成组织悬液,然后将组织悬液在-20?条件下反复冻融三次,最后进行RNA的抽提。
2.2 抽取RNA
每100mg组织匀浆加1mlTrizol试剂,样品体积不能超过Trizol试剂的10%。
1)、 取病料组织匀浆100mg置研磨器中剪碎并研磨,加入Trizol试剂1ml继续研磨。
1)、 尿囊液100ul加900ulTrizol试剂,剧烈振荡30秒,室温放置10min。
2) 、将组织匀浆液转移至1.5ml离心管中,室温放置5min,
3)、 12000 rpm 4?离心10 min,取上清;
4) 、按每2ml匀浆物中加200ul氯仿/异戊醇(24:1)混合液,剧烈振荡30秒;室温放置15min ,使核蛋白复合体彻底裂解;
5)、 12000 rpm 4?离心15 min
6)、 小心吸取上层水相 ,不要吸取水相的最下层,为了降低被处于水相和有机相分界处的DNA污染的可能性。
7)、 加入等体积异丙醇,混匀,4?放置至少10min,
8)、 12000 rpm 4?离心15 min;RNA应该在管底形成黄/白色的沉淀。
9)、 小心弃上清,按1ml75%酒精/1ml Trizol试剂比例加-20?预冷的75%酒精,通过摇晃或吹打使RNA
沉淀重悬;
10)、 12000 rpm 4?离心8 min;
11)、 吸头吸弃上清,室温下放置5-5min使酒精挥发(不能完全干燥);
12)、 用30ul DEPC水溶解沉淀,通常先将样品冷冻后融化对增加样品的可溶性有帮助。
13)、 病毒RNA于-20?保存备用或直接用于反转录。
[注意]
1)、整个操作要带口罩及一次性手套,并尽可能在低温下操作。
2)、提取上清这步一定要小心,靠近沉淀的部分一定要舍得不要,要不然会有蛋白质污染,影响比值 3)、在所有RNA实验中,最关键的因素是分离得到全长的RNA。而实验失败的主要原因是核糖核酸酶(RNA酶)的污染;RNA酶可耐受多种处理而不被灭活,如煮沸、高压灭菌等;
4)、研究人员造成的污染 RNA酶最主要的潜在污染源是研究人员的手。因此进行RNA实验时应勤换手套。 5)、试验过程中,模板中含有杂蛋白质,?模板中含有Taq酶抑制剂,?模板中蛋白质没有消化除净,特别是染色体中的组蛋白,?在提取制备模板时丢失过多或吸入酚都会导致假阴性结果,不出现扩增条带。 6)、常用的RNA酶抑制剂:
(1). 焦磷酸二乙酯(DEPC):是一种强烈但不彻底的RNA酶抑制剂。它通过和RNA酶的活性基团组氨酸的咪唑环结合使蛋白质变性,从而抑制酶的活性。
(2). 异硫氰酸胍:目前被认为是最有效的RNA酶抑制剂,它在裂解组织的同时也使RNA酶失活。它既可破坏细胞结构使核酸从核蛋白中解离出来,又对RNA酶有强烈的变性作用。
7)、介绍一个可以确认RNA溶液中有没有残留的RNA酶的方法:
保温试验
方法很简单的,按照样品浓度,从RNA溶液中吸取两份1000 ng的RNA加入至0.5 ml的离心管中,并
且用pH7.0的Tris缓冲液补充到10 ul的总体积,然后密闭管盖。把其中一份放入70?的恒温水浴中,保温
1 h。另一份放置在-20?冰箱中保存1 h。
时间到了之后,取出两份样本进行电泳。电泳完成后,比较两者的电泳条带。如果两者的条带一致或
者无明显差别(当然,它们的条带也要符合方法2中的条件), 则说明RNA溶液中没有残留的RNA酶污
染,RNA的质量很好。相反的,如果70?保温的样本有明显的降解,则说明RNA溶液中有RNA酶污染。 3 紫外分光光度计检测RNA纯度
做RT前必需测RNA浓度,逆转录体系对RNA量还是有一些要求,常用500ng或1ug。
取10ul抽取的IBV RNA液,加入灭菌的DEPC水至3ml,用紫外分光光度计测量样品在230nm、260nm和280nm处的吸光度,并用OD260/OD280和OD260/OD230值,估算RNA的纯度。
纯的RNA的OD260/OD280应为2.0,高于2.0表明有RNA降解,低于2.0表明有蛋白质污染。而 OD260/OD230应大于2.0 ,若OD260/OD230小于2.0表明有残存的酚、多糖、胍盐的污染。 4 电泳检测RNA的完整性
RNA也最好至少要用电泳,一方面定量,另一方面可以看看完整性。
取DEPC水处理过的PCR管,加入RNA为5.5ul,37%甲醛3.5ul,甲酰胺10 ul,10×甲醛电泳缓冲液1 ul,放入PCR仪中65?进行变性处理15min。迅速冰浴冷却,离心后,再加入2 ul RNA加样缓冲液。
电泳液为1×甲醛电泳缓冲液,1%琼脂糖甲醛凝胶,70-80V 5 min预电泳,50-60V2-2.5 h 电泳(溴酚蓝至约8cm电泳结束)。紫外光下观察电泳结果并拍照。
5 cDNA合成(RT)
反转录选择20ul 反应体系,冰盒上操作。
1)、微量管中加6ulRNA加2ul反转录引物,混匀,65?,5 min
2)、立即冰浴冷却(2 min),稍离心
3)、继续加入以下组分
5×反转录反应缓冲液 4 ul
dNTP(2.5mmol/L) 4 ul
反转录酶(200U/ul) 1 ul
RNA酶抑制剂(40U/ul) 0.5 ul
DEPC水 2.5 ul
4)、混匀,42?水浴1h,接着95?变性10min(破坏反转录酶),冰上冷却5min.
5)、直接进行PCR,或放于-20?保存备用。
6)、试验同时设阴性对照和阳性对照。
注意
1)、如果用PCR仪来控制温度和时间的, RT需在PCR反应管中作。
2)、步骤(4)中将RNA溶液置65?中温育然后冷却至室温再上样的目的有两个,一个是破坏RNA的二级结构,尤其是mRNA Poly(A )尾处的二级结构,使Poly(A )尾充分暴露,从而提高Poly(A )RNA的回收率;另一个目的是能解离mRNA与rRNA的结合,否则会导致rRNA的污染。所以此步骤不能省略。
6 N基因目的片断的PCR扩增
PCR反应液配制:按50 ul反应体积配制,按PCR操作说明书加样。
(1)准备主混合物,根据样品个数按以下浓度加入各成分
dNTP(2.5mmol/L) 4 ul
Taq DNA聚合酶缓冲液10× 5 ul
MgCl(25mmol/L) 3 ul 2
Taq DNA聚合酶(5U) 0.2 ul
上游引物 2.0 ul
下游引物 2.0 ul
cDNA(反转录产物) 2.5 ul
DEPC水 31.3 ul
(2)混合,稍离心(让反应物下沉官底),铺上50ul矿物油
(3)根据下列反应条件进行PCR循环:
先94?3min,
再 94?40s、
59?30s、 共30个循坏
72?30s,
最后72?7min,最终产物可以行琼脂糖电泳。
注意:PCR反应的关键环节有?模板核酸的制备?引物的质量与特异性,?酶的质量?PCR循环条件。 1)反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul或100ul,应用多大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20ul 后,再做大体积时,一定要模索条件,否则容易失败。
2)加样:原则是DNA最后加,其他试剂按照体积大小从大往小的加。操作多份样品时,算出总体积后在一个管子里面制备反应混合液,先将dNTP、缓冲液、引物和酶混合好,然后再分装到各个管子里,这样既可以减少操作,避免污染,又可以增加反应的精确度。必要时,在加标本前,反应管和试剂用紫外线照射,以破坏存在的核酸。
3)酶及其浓度 目前有两种Taq DNA聚合酶供应, 一种是从栖热水生杆菌中提纯的天然酶,另一种为大肠菌合成的基因工程酶。催化一典型的PCR反应约需酶量2.5U(指总反应体积为100ul时),浓度过高可引起非特异性扩增,浓度过低则合成产物量减少。
4)dNTP的质量与浓度和PCR扩增效率有密切关系,dNTP粉呈颗粒状,如保存不当易变性失去生物学活性。dNTP溶液呈酸性,使用时应配成高浓度后,以1M NaOH或1M Tris。HCL的缓冲液将其PH调节到7.0,7.5,小量分装, -20?冰冻保存。多次冻融会使dNTP降解。在PCR反应中,dNTP应为50,200umol/L,尤其是注意4种dNTP的浓度要相等( 等摩尔配制),如其中任何一种浓度不同于其它几种时(偏高或偏低),就会引起错配。浓度过低又会降低PCR产物的产量。dNTP能与Mg2+结合,使游离的Mg2+浓度降低。 5)Mg2+浓度 Mg2+对PCR扩增的特异性和产量有显著的影响,在一般的PCR反应中,各种dNTP浓度为200umol/L时,Mg2+浓度为1.5,2.0mmol/L为宜。Mg2+浓度过高,反应特异性降低,出现非特异扩增,浓度过低会降低Taq DNA聚合酶的活性,使反应产物减少甚至使PCR扩增失败而不出扩增条带。 6)变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出现假阴性;退火温度过低,可致非
特异性扩增而降低特异性扩增效率退火温度过高影响引物与模板的结合而降低PCR扩增效率。有时还有必要用标准的温度计,检测一下扩增仪或水溶锅内的变性、退火和延伸温度,这也是PCR失败的原因之一。 7) PCR反应条件为温度、时间和循环次数。
温度与时间的设置:基于PCR原理三步骤而设置变性-退火-延伸三个温度点。在标准反应中采用三温度点法,双链DNA在90,95?变性,再迅速冷却至40 ,60?,引物退火并结合到靶序列上,然后快速升温至70,75?,在Taq DNA 聚合酶的作用下,使引物链沿模板延伸。对于较短靶基因(长度为100,300bp时)可采用二温度点法, 除变性温度外、退火与延伸温度可合二为一,一般采用94?变性,65?左右退火与延伸(此温度Taq DNA酶仍有较高的催化活性)。
?变性温度与时间:变性温度低,解链不完全是导致PCR失败的最主要原因。一般情况下,93?,94?min足以使模板DNA变性,若低于93?则需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或PCR产物完全变性,就会导致PCR失败。
?退火(复性)温度与时间:退火温度是影响PCR特异性的较重要因素。变性后温度快速冷却至40?,60?,可使引物和模板发生结合。由于模板DNA 比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱基组成及其浓度,还有靶基序列的长度。对于20个核苷酸,G+C含量约50%的引物,55?为选择最适退火温度的起点较为理想。引物的复性温度可通过以下公式帮助选择合适的温度:
Tm值(解链温度)=4(G+C),2(A+T)
复性温度=Tm值-(5,10?)
在Tm值允许范围内, 选择较高的复性温度可大大减少引物和模板间的非特异性结合, 提高PCR反应的特异性。复性时间一般为30,60sec,足以使引物与模板之间完全结合。
?延伸温度与时间:
Taq DNA聚合酶的生物学活性:
70,80? 150核苷酸/S/酶分子
70? 60核苷酸/S/酶分子
55? 24核苷酸/S/酶分子
高于90?时, DNA合成几乎不能进行。
PCR反应的延伸温度一般选择在70,75?之间,常用温度为72?,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1Kb以内的DNA片段,延伸时间1min是足够 的。3,4kb的靶序列需3,4min;扩增10Kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。
7 琼脂糖凝胶电泳检测
(1)将倒胶槽两端用透明胶带封闭并放置在水平的台面上,放好梳板,使梳板齿下沿距倒胶槽板1mm. (2)配1.0%琼脂糖凝胶
1g琼脂糖加100ml 1×TAE电泳缓冲液,加热煮沸后以蒸馏水补足体积至100ml,迅速混匀,待冷却至60?左右时,加入100ul 0.5mg/ml的EB液,充分混匀。
(3)倒胶至倒胶槽内,使厚度为3-5mm,排除气泡后待胶完全凝固,撕去两端胶带。
(4)将倒胶槽置电泳槽中,加入1×TAE电泳缓冲液,使其没过胶面2-3mm,轻轻拔出梳板 (5)加样:取2ul上样缓冲液于Parafilm膜上,加PCR产物5ul,混匀后,加入点样孔,不要溢出孔外,同时设DNA分子量标准。
(6)电泳:样品在负极端,接通电源,130V,100mA,20 min.
(7)电泳结束后,关闭电泳仪,取出倒胶槽,将电泳完毕的琼脂糖凝胶于紫外凝胶成像系统计录结果 (8)结果判定:在阳性对照孔出现相应扩增带,阴性对照孔无扩增带时判定结果。
若样品扩增带与阳性扩增带处于同一位置,则判定IBV阳性,否则为阴性。
注意:PCR产物的电泳检测时间一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。
范文二:pcr反应模板的制备
PCR反应模板的制备
PCR反应的关键因素主要有引物的选择与设计,酶的质量.模板的制备,在前二者都稳定可行的情况下,PCR模板的制备尤为重要.模板处理方法的选择及操作人员的基 本技能,决定分离模板核酸(DNA或RNA)的质和量及PCR的成败.而提高模板核酸质量的 关键是除去杂质(蛋白质、酶、脂肪等).除去抑制Taq DNA聚合酶活性抑制因子,提高模板核酸的产量.
传统的核酸模板提取方法是采用去垢剂如SDS等来破坏细胞组份,溶解细胞膜, 使蛋白质变性,再用蛋白酶K来消化去除蛋白质,尤其是与DNA结合的蛋白质,再用 酚:氯仿抽提,然后用乙醇或异丙醇沉淀核酸,供PCR实验用.但近几年来也发展了些 简便实用较为有效的标本消化处理方法.亦可满足PCR实验的要求.采用哪种方法消化 处理标本,视PCR实验的目的(是科研还是检测)及环境条件而定.
1微克人基因组DNA相当于3*10的五次方靶分子,
10纳克酵母DNA相当于3*10的五次方靶分子
1纳克大肠杆菌DNA相当与??????
所以,扩增不同拷贝数的靶序列时,加入含靶序列的DNA量也不同
如真核核糖体RNA基因有200~500拷贝,反应中仅需加入0.5~2纳克人基因组DNA即可
以质粒DNA或染色体DNA为模板时扩增最适条件不同。前者所需的酶量少,循环数少,温度不如染色体DNA要求严格。 模板核酸的提取制备方法
(一)蛋白酶K消化裂解法:适用于所有标本的消化处理,尤以DNA样品为佳.如组 织细胞(包括石蜡包埋组织)绒毛、毛发、精斑、血液(血清、血浆、全血)局部分泌 物、尿,粪便等.
1.试剂
?蛋白酶K消化液:10mmol/L Tris.cl(PH8.0)
10mmol/LEDTA
150mmol/LNaCL
0.5%SDS
100,200ug/ml蛋白酶K.
?蛋白酶K最好用水配成20mg/ml,临用时加入消化液中.
2.提取方法:
有些标本、在用蛋白酶K消化前,还需预处理一下,如粪便、分泌物、痰液、组 织块、石蜡包埋组织等,其方法有离心去掉杂质,脱蜡等.
临床标本或经预处理的标本加蛋白酶K裂解液50,100ul.混匀,55?1,3小时, 或37?过夜.加等体积的饱和酚抽提1,2次,再加等体积的氯仿:异戊醇(49:1)抽提一 次,上清加入1/10体积的pH值5.23mmol/L醋酸钠缓冲液,加入2.5倍体积的冰冷无水 乙醇(或加等体积的异丙醇)-20?放置至少3h.取出后14000转/min离心15min.小心吸 弃或倒出上清,沉淀加入75%冰冷乙醇15000r/min5min离心洗涤1,2次,特别小心的 吸弃或倒掉上清,真空或37?温箱或室温干燥,加TE缓冲液20ul.溶解后,取3,8ul 用于PCR扩增,或放-20?保存.
蛋白酶K消化法除上述经典处理法外,亦可在蛋白K消化处理标本,经离心处理 后,吸取上清经95,97?或煮沸10min灭活蛋白酶K后,直接作核酸模板用于PCR扩增. 如杂质较多,还可经酚:氯仿抽提后,即可用于PCR反应.此法蛋白质及其它杂质消除 彻底,Taq酶活性不受影响,具有良好的重复性与稳定性.但操作繁复,技术要求高.
(二)直接裂解法:标本(组织细胞,分泌物)加PBS或生理盐水离心洗涤后,加消化 裂解液20,50ul(0.5%NP-40和0.5%吐温-20),95,98?,15,30min以裂解病原体, 裂解细胞.然后
12000r/min离心5,10min,取上清20,30ul用于PCR扩增.血清标本可 直加等体积的消化液,加热处理离心后PCR扩增.亦有用5%的NP-40和1.5%2-ME做裂解 液,95?30min消化处理,离心取上清,PCR扩增检测HBV DNA.
(三)碱变性法:取血清20ul,加入1mol/L NaOH 20ul,37?30min,离心,加 1mol/L HCl 20ul,离心后,取上清5ul,用于PCR扩增.*亦有用10ul血清加NaOH至 0.2mol/L,37?1h,再加HCL中和离心后取上清10ul做PCR,其特异性和敏感性较前法 更好.
(四)煮沸法:经离心洗涤过的组织细胞,分泌物及血液标本,加适量无菌蒸馏水或PBS,混匀 后,100?煮沸10,15min,14000r/min 10min,取上清做PCR.
(五)碘化钠法:取组织细胞及抗凝血液10,100ul,加等体积的6MNaI,反复轻混 20s,加等体积氯仿:异戊醇(49:1)振匀后,离心取上清加0.6体积的异丙醇,混匀后 置室温3min.14000r/min 10min,沉淀加10,100ul,TE溶解,取5,10ul做PCR,亦有 用100ul血清加裂解液300ul(6M NaI,0.5%十二烷基肌酸钠,10ug糖原,26mmol/L pH8.0 Tris-HCL,13mmol/LEDTA)混匀后,60?15min,加等体积异丙醇,离心沉定 后,加TE液用于PCR扩增,其产量和质量较高.
(六)异硫氰酸胍法
此法主要用于RNA的提取.标本为组织细胞及血清等.
1.异硫氰酸胍消化液
异硫氰酸胍4mol/L
柠檬酸钠(PH7.0)25mmol/L
十二烷基肌酸钠0.5%
β-巯基乙醇0.1mol/L
2.消化方法:用50,100ul细胞悬液及血清,加等体积的异硫氰酸胍消化液振混匀 后,或65?1小时,或室温放置数分钟,然后加1/10体积的3mmol/L pH5.2的醋酸钠缓 冲液,再加等体积的酚:氯仿,用力振摇约10s,10000r/min,离心5min,取上清加等 体积异丙醇-20?放置3h后,14000r/min离心15min,沉定用75%冰冷乙醇离心洗涤1, 2次,真空干燥,沉淀用TE缓冲液溶解即可用于逆转录PCR扩增,或放-20?保存.
其它消化处理标本提模板核酸的方法有?异硫氰酸胍一二氧化硅法?异硫氰酸胍 -玻璃粉
法.?Chelex-100法.?全血直接扩增法?尿素消化裂解法.各有千秋,可根据 情况选用.
特殊组织DNA的提取及鉴定
1、提取DNA的简易方法:
1)、从全血中提取DNA:取20μl抗凝血于0.5ml无菌管中,加500μl左右的ddH2O混匀后,
12000rpm离心2分钟,弃上清,(若沉淀中仍 有红细胞,需重复此操作1-2次)加入样品提取液20μl,混匀,100?煮10分钟(可在扩增仪上进行)后12000rpm离心5分钟,取上清2μl进 行扩增。
2)、从其它有核细胞中提取DNA:可直接加适量的样品提取液到装有发根、头屑、骨髓、精
子、组织碎片等有核细胞的管中,100?煮10分钟(可在扩增仪上进行)后12000rpm离心5分
钟,取上清进行扩增。
2、从全血中用有机溶剂提取DNA:
1)试剂
a.10%SDS
b.2.0M醋酸钠
c.20mg/ml蛋白酶K
d.苯酚:氯仿:异戊醇(25:24:1)
e.分析纯乙醇
f.TE(10mM Tris,1.0mMEDTA溶液,Ph7.5) 2)步骤
A、血液样本收集于含EDTA的抽血用管子中,样品用手倒转试管混合后等分。每份700μl全
血可放在1.5ml的塑料离心管中,-70?储存。
B、在解冻的(或液体的)血液中加入800μl的TE并混匀,12000rpm离心2分钟。
C、吸取1.0ml上清液并弃去。
D、加入1.0mlTE,振荡后离心2分钟,去除尽可能多的上清液,但不要影响沉淀。
E、向沉淀中加入:375μl 2M醋酸钠; 25μl 10%SDS;5μl 蛋白酶K溶液。略加振荡,于5
6?保温一小时。
F、加入120μl苯酚/氯仿/异戊醇,振荡30秒。 G、12000rpm离心5分钟。
H、小心移出水相(上层)到一新的1.5ml的离心管中。 I、向水相中加入2.5倍体积预冷的分析纯乙醇,倒转试管加以混合,将试管于-20?下放置
10分钟以上。
J、12000rpm离心10分钟。
K、倾去酒精,用移液器小心吸去剩余酒精,不要触动沉淀。 L、加180μl TE 后振荡。加20μl2M醋酸盐,手工混匀。
M、加500μl预冷分析纯酒精,用手轻轻混匀,12000rpm离心10分钟,倾去上清液。
N、用室温下的70%酒精1ml洗涤DNA沉淀,12000rpm离心5分钟,倾去上清液。
O、用移液器吸去残余的酒精,管子置于真空离心抽除残余的酒精(大约10分钟)。或将管
子放在100?以下烘干。
P、加200μl TE于DNA沉淀中,混匀后于4?保温过夜(或56?溶解DNA 2小时以上),次日
清晨振荡10秒。
3、从斑痕材料中用有机溶剂提取DNA
1)试剂
a.斑痕抽提缓冲液(1.21g Tris,5.84gNaCl,6.02g二硫苏糖醇, 2%SDS,10mMEDTA/升,pH8.0)
b.其它试剂见2.1)
插入:血液的模板制备:取0.2毫升抗凝血液注入1.5ml离心管中,加1毫升细胞裂解缓冲液
c细胞裂解缓冲液:0.32mol/L蔗糖
5摩尔每升的氯化镁,
1%TritonX-100
0.01mol/L Tris.HCl
混匀,静置10分钟,3000g离心十分钟,弃上清,沉淀加50微升DNA提取缓冲液:50毫摩尔每升的氯化钾
2.5毫摩尔每升的氯化镁
1毫克每毫升的明胶
0.5%NP-40
0.5%Tween-20
200微克每毫升的蛋白酶K
10毫摩尔每升的Tris.HCl
pH8.0 65摄氏度水浴1小时,12000g离心十分钟,弃沉淀,上清直接进行PCR反应
2)步骤
A、将斑痕剪成碎片后置于1.5ml的塑料离心管中。 B、加400μl斑痕抽提缓冲液及10μl蛋白酶K溶液,混匀,离心数秒使碎片浸入液体。
C、于56?保温过夜。
D、用一新的灭菌牙签将碎片中的液体拧干后从管中取出。 E、加500μl苯酚/氯仿/异戊醇,手摇混匀,12000rpm离心3钟。
F、将上清液转移到新的离心管。
G、上清液中加入2.5倍体积预冷的分析纯酒精。 H、手摇混匀试管中的各种成分,放置-20?10分钟以上。 I、12000rpm离心10分钟。
J、倾去酒精。
K、用1ml室温的70%酒精洗涤。
L、12000rpm离心5分钟。
M、倾去酒精,用移液器吸去残余酒精。
N、真空离心10分钟除去残余酒精,或在100?以下烘干. O、56?下用36μl TE溶解DNA,时间至少2小时。 4、从精斑中分离提取DNA
1)试剂
a.TNE缓冲液(1.21g Tris,5.84gNaCl,0.37gEDTA/升) b.20%sarcosyl
c.0.39M二硫苏糖醇
d.其它试剂见2.1)
2)步骤
A、从棉签上取下有检材的棉花并置于1.5ml塑料离心管中。 B、管中加入400μl TNE缓冲液;25μl 20%sarcosyl;75μl水;5μl蛋白酶K溶液。手摇混
匀,37保温2小时。
C、用一新的灭菌牙签将棉花拧干后从管中取出,再用12000rpm离心5分钟。
D、移上清液(内含裂解细胞或"雌性"片段)于一新的1.5ml管,不要振荡沉淀物(内含非
裂解细胞或精子)。再用TNE洗脱沉淀物四次。 E、在有沉淀的管中加入
10μlTNE;50μl20%sacrosyl;40μlDTT;150μl水;10μl蛋白酶K
溶液。手摇混匀,37?保温2小时。
E,O的方法分别抽提出两管中的雌性DNA和精子F、按3,2)
DNA。
5、用Chelex从全血/血斑中抽提DNA
1)试剂
a.TE
b. 5%Chelex 100(w/v于无菌水中)
2)步骤
A、3μl全血或3mm×3mm的血斑置于灭菌的1.5ml离心管中,加入灭菌的1mlTE于管中,振荡
数秒。
B、置于室温30分钟,振荡数秒。
C、用12000rpm离心3分钟。
D、弃上清液,保留足够上清液盖没沉淀,勿搅起沉淀。 E、加入200μl 5%Chelex,振荡数秒。
F、于56?保温30分钟。
G、振荡数秒。
H、沸水浴8分钟。
I、振荡数秒。
J、用12000rpm离心5分钟,上清中为提取的DNA。 6、用Chelex从精斑中抽提DNA
1)试剂
a.TE
b.10mg/ml蛋白酶K
c.20% Chelex100(w/v于无菌水中)
d.洗脱缓冲液(10mM Tris,10mM EDTA,50mMNaCl,2%SDS,Ph7.5)
e.1M二硫苏糖醇
2)步骤
A、将附有检材的棉签或纤维置于1.5ml灭菌离心管中,加1ml灭菌的TE,振荡数秒。
B、室温30分钟。
C、振荡数秒。
D、用一新的灭菌牙签将残片拧干后从管中取出。 E、用12000rpm离心3分钟。
F、弃去上清液,但保留约50μl上清液,勿使"细胞沉淀"搅起(若沉淀体积较大,应保留
更多上清液,在此例中,保留的上清液体积应为沉淀的2倍)。 G、加150μl灭菌的蒸馏去离子水和2μl蛋白酶K溶液。
H、56?保温1小时以裂解非精子细胞。
I、用12000rpm离心5分钟,沉淀为"精子沉淀"。 J、取150μl上清液(含裂解细胞组分)及到一新的1.5ml离心管中,再加50μl 20%Chele
x,振荡数秒后,稍加离心,然后接步骤O,S进行操作。 K、用0.5ml洗脱缓冲液重新悬浮原管中的沉淀,略加振荡, 用12000rpm 离心5分钟,弃去
约450μl上清液。
L、重复步骤K再洗四次。
M、用1ml灭菌的蒸馏去离子水重新悬浮沉淀,稍经振荡,用12000rpm离 心5分钟,弃去
950μl上清液。
N、加150μl的5%Chelex,2μl蛋白酶K溶液,7μl 1M的二硫苏糖醇 于"精子组分",振
荡数后,稍加离心。
O、56?保温1小时。
P、振荡各管数秒。
Q、样品于沸水浴8分钟。
R、振荡数秒。
S、用12000rpm离心3分钟,
绒毛:绒毛样品离心2分钟,弃上清,沉淀用TE缓冲液洗二次
悬于50微升裂解液中强烈震荡,煮沸2分钟,;离心后取
上清进行PCR扩增。
范文三:pcr步骤
一、知识背景 :
1、基因表达:DNA RNA Protein
单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因 104个丝心蛋白 mRNA (每个 mRNA 存活 4d , 可以合成 105个丝心蛋白) 共合成 109个丝心蛋白。 因此单拷贝基因的 mRNA 表达水平 对于其功能水平的调控是非常重要的。
合成的引物序列决定。反应分三步:
A 、变性:通过加热使 DNA 双螺旋的氢键断裂,形成单链 DNA ;
B 、退火:将反应混合液冷却至某一温度,使引物与模板结合。
C 、延伸:在 DNA 聚合酶和 dNTPs 及 Mg2+存在下,退火引物沿 5' 3'方向延伸。
以上三步为一个循环,如此反复。
3、逆转录酶和 RT-PCR
逆转录酶(reverse transcriptase)是存在于 RNA 病毒体内的依赖 RNA 的 DNA 聚合酶,至少具有 以下三种活性:
1、依赖 RNA 的 DNA 聚合酶活性:以 RNA 为模板合成 cDNA 第一条链;
2、 Rnase 水解活性:水解 RNANA 杂合体中的 RNA ;
3、依赖 DNA 的 DNA 聚合酶活性:以第一条 DNA 链为模板合成互补的双链 cDNA.
二、 RT-PCR 的准备 :
1、引物的设计及其原则:
1)引物的特异性决定 PCR 反应特异性。因此引物设计是否合理对于整个实验有着至关重要的影响。 在引物设计时要充分考虑到可能存在的同源序列,同种蛋白的不同亚型,不同的 mRNA 剪切方式以
及可能存在的 hnRNA 对引物的特异性的影响。尽量选择覆盖相连两个内含子的引物,或者在目的蛋 白表达过程中特异存在而在其他亚型中不存在的内含子。
2)引物设计原则的把握:引物设计原则包括
a 、引物长度 :一般为 15~30bp ,引物太短会影响 PCR 的特异性,引物太长 PCR 的最适延伸温度会 超过 Taq 酶的最适温度,也影响反应的特异性。
b 、碱基分布:四种碱基最好应随机分布,避免嘌呤或嘧啶的聚集存在,特别是连续出现 3个以上的 单一碱基。 GC 含量 (Tm 值) :40%~60%, PCR 扩增的复性温度一般是较低 Tm 值减去 5~10度。 c 、 3' 端要求:3' 端必须与模板严格互补,不能进行任何修饰,也不能有形成任何二级结构的可能。 末位碱基是 A 时错配的引发效率最低, G 、 C 居中间,因此引物的 3' 端最好选用 A 、 G 、 C 而尽可能 避免连续出现两个以上的 T 。
d 、 引物自身二级结构:引物自身不应存在互补序列, 否则会自身折叠成发夹状结构或引物自身复性。
e 、引物之间的二级结构:两引物之间不应有多于 4个连续碱基互补, 3' 端不应超过 2个。
f 、同源序列:引物与非特异扩增序列的同源性应小于连续 8个的互补碱基存在。
g 、 5' 端无严格限制:5' 末端碱基可以游离,但最好是 G 或 C ,使 PCR 产物的末端结合稳定。还可以 进行特异修饰(标记、酶切位点等)等等。
根据实验目的选择适当的引物。 常用引物设计软件如 Primer5.0, Oligo6.0等对于这些条件都可以自 行设置。
处理,除去 RNA 酶,防止操作过程中 RNA 降解。然后经高压灭活(灭菌和灭活 DEPC )。
3、 试剂 准备:变性液、水饱和酚、乙酸钠、氯仿、异丙醇、 75%酒精、经 DEPC 处理并高压的水。 三、 RNA 的提取方法:
RT -PCR 中从细胞分离的 RNA 的质量至关重要, 包括 RNA 的纯度和完整性。 RNA 分离的最关键因
素是尽量减少 RNA 酶的污染。但 RNA 酶活 性非常稳定,分布广泛,除细胞内源性 RNA 酶外,环 境中也存在大量 RNA 酶。因此在提取 RNA 时,应尽量创造一个无 RNA 酶的环境,包括去除外源性 RNA 酶污染和抑制内源性 RNA 酶活性,主要是采用焦碳酸二乙酯(DEPC )去除外源性 RNA 酶, 通过 RNA 酶的阻抑蛋白 Rnasin 和强力的蛋白质变性剂如异 硫氰酸胍抑制内源性 RNA 酶。
实验操作步骤
一、实验器具与材料:
1、移液枪:1ml 、 200μl 、 20μl 、 10μl 、 2μl
3、匀浆管:
5ml
5、 EP 管:1.5ml 、 0.2ml 、 100μl
6、 试剂 瓶:2个 60ml 的棕色试剂瓶(广口,带盖)
1个 125ml 的白色试剂瓶(放无水乙醇)
10、盐水瓶:250ml 、 500ml 各 2个备用,一个装无水乙醇,另一个装 DEPC 水
11、铝制饭盒:4个
12、塑料小饭盒:1个
13、大瓷缸:2个
RT-PCR 原理与实验操作步骤
14、锡泊纸:一卷
15、卷纸:2卷
16、三角烧瓶:带盖,稍大
二、实验器具的处理与准备
1、塑料制品:(包括枪头、 EP 管、匀浆管等)
水,过夜,然后高压,再烤干备用,实验前将枪头等放入吸头台,再高压一次(EP 管)
2、玻璃制品:泡酸过夜,冲洗干净,蒙锡纸烤干备用(DEPC 水泡)(洗净后先泡 1‰ DEPC 过夜, 再烤干)
3、匀浆器:(包括剪刀、镊子)先洗净后,再高压(不需要泡 DEPC )
三、试剂配制:
1、 DEPC 水:吸出 1ml 放在 1000ml 双蒸水中配成 1‰ DEPC 水,放在 1000ml 容量瓶中静置 4小 时备用。
2、 75%乙醇:用无水乙醇 DEPC 水配,然后放 -20℃保存(其中 DEPC 水需先高压)
3、异丙醇:放入棕色瓶中
4
、氯仿:放入棕色瓶中
四、几种缓冲液的配制:
1、电泳缓冲液:
硼酸 27.5g
蒸溜水 1000ml
5×TBE (贮存液)
再将 5×TBE 稀释 10倍成 0.5TBE 就可以在电泳时使用 (即工作液浓度) , 如取 50ml 贮存液 450ml 水 --→ 500ml 工作缓冲液
2、上样缓冲液:
0.25%溴酚蓝
0.25%二甲苯青 FF
RT-PCR 原理与实验操作步骤
30%甘油
6×缓冲液, 4℃保存
1、 1.0%:
1.0g 琼脂糖 100ml 电泳缓冲液,微波炉中火 30秒至沸腾,熔化的琼脂物冷却至 60℃时加入 10mg/ml溴化乙锭 2.5μl ,充分混匀,将温热的凝胶倒入已置好梳子的胶膜中,在室温下放置 30-45min 后现进行电泳。
2、 1.5%:
同上,将琼脂糖的量改为 1.5g
范文四:pcr实验步骤
1.只写一份实验报告,写成论文的形式,不必写预习报告(如果一张实验报告不够的话,可以再附一张实验报告)。
2.实验报告的主题是关于“转基因食品的检测”,大家可以查阅相关的资料,然后结合实验课上的方法、步骤、结果。
3.实验报告大致可以分为“前言”、“方法”、“结果与讨论”三个板块。
4.老师曾要求写过的“实验总结”可以写在“前言”板块中。
5.本次实验报告虽然就只有一份 ,但是算作实验考试成绩,请亲们认真对待。
目前,转基因存在的检测方法主要有prc(凝合酶链反应)技术、凝胶电泳测序和elisa(酶联免疫法)等,这些方法在食品和医药研究方面都有广泛的使用,其中以prc技术最为成熟。该技术是由美国centus公司的karymullis发明,于1985年由saiki等首次报道,是近年来开发的体外快速扩增dna技术。1988年,r.k.saiki等人又成功地将热稳定taqdna聚合酶应用于pcr扩增,是pcr技术上向实用化的一次突破性的进展。通过pcr技术可以简便快速地从微量生物材料中以体外扩增的方式获得大量特定的核酸,并且有很高的灵敏度和特异性。pcr既可做定性又可做定量分析,目前大多以定性检测为主,定性检测的检出范围为百分之0.1。但该项技术的检测结果也有可能与实际不相符合,会出现假阴性或假阳性结果(即检测物质本身含有转基因物质,而未被检出;或是本身没有转基因物质,而检出有转基因成分)。
由于许多获得批准的遗传工程体(gmo)表现出一定的导入基因的性状,因此,基因检测也需要以蛋白质为基础的检测技术,这就出现了elisa法。1971年engvall和perlman发表了有关酶联免疫吸附分析法用于免疫球蛋白g(igg)定量测定的文章,使该技术发展成为液体标本中微量物质的测定方法。elisa法与pcr法相比具有操作简单、快速、结果准确,有高通量的特性,解决了转基因检测中样品核酸制备的困难,同时降低了检测成本和时间高的催化效率,可极大地放大反应效果,从而使测定方法达到很高的灵敏度和稳定性。但该项技术主要应用于原料和半成品分析,在终产物分析方面灵敏度底于pcr法。另外,该项技术还需要特殊的抗体和“新”的表达蛋白,而食品中的这类“新”蛋白质的含量极低,且在加工过程中蛋白质常会发生变化;该分析法同时还需要熟练的操作技术。这些因素都使elisa法在转基因检测应用上受到了一定的限制。
外先进经验,尽快制定有关卫生行政许可司法审查和司法救济
依赖保护的法律体系,减少错误、无效的行政许可,迫使卫生行
政机关和工作人员依法行政。
316 加大宣传教育力度,增强民众的自我保护意识 通过多
种渠道如新闻媒体、电子触摸屏、互联网、政务公示栏,宣传《中
华人民共和国食品卫生法》、《行政许可法》、《卫生行政许可管
理办法》等相关卫生法律法规、行政许可工作流程等,切实加强
对管理相对人的卫生法律法规知识的培训,增强民众的法治观
念,树立法律的最高权威,让民众拿法律作武器,增强其自我保
护意识。
参考文献
[ 1 ] 卫生部. 卫生行政许可管理办法. 中华人民共和国令,第38 号. [ 2 ] 卫生部. 关于卫生监督体制改革的意见. 卫办发[ 2000 ]第16 号. [3 ] 张岩,薛清春,刘静,刘培国. 项目管理运用于疾病控制领域的探
索与体会. 中国公共卫生管理,2006 ,22 (1) :15 - 16. . [ 4 ] 陈汉平,吴涛. 加强卫生监督机构信息管理的几点体会. 中国公共 卫生管理,2006 ,22 (1) :74 - 76.
(收稿日期:2007 - 04 - 17)
转基因食品检测技术探讨
施向东 林新勤 梁惠宁 龙兮 (南宁市疾病预防控制中心,530011)
中图分类号:R155. 5 文献标识码:A 文章编号:1001 - 9561 (2008) 01 - 0049 - 02
由转基因生物转化产生的食品为转基因食品。转基因食 品英文原译名为基因修饰食品( Genetically Modified Food ,简称 GMF) ,指利用基因工程技术改变基因组后构成的动物、植物 和微生物生产出的食品和食品添加剂。它将会使我们产生一 个全新的食品概念并使饮食文化发生新的变化。现在社会对 其的忧虑有二点:通过生物技术产生的新食品成分、食品添加 剂对人体健康和生长发育是否有潜在的负面影响;基因食品中 的新因基通过食物链和环境释放等途径是否影响自然界生物 多样性。虽然迄今为止尚未发现有证据的转基因食品对健康 和环境的危害,但由于转基因食品的安全性评价具有累积性和 潜在性特点,并与社会、文化及伦理等多方面因素互为影响,所 以需要多方位长期系统的监测才能对其安全性作出较为客观 的评价,转基因食品的检测技术在其中尤为重要。国际上对转 基因作物、食品安全性评价适宜方法和策略达成初步一致,评 价原则主要包括三个层次:了解被评价食物的遗传背景与基因 改造方法,检测食品中可能存在的毒素、过敏源等,进行毒理学 实验,推荐对转基因食品的安全性评价应用“实质性等同”原 则[ 1 ] ,考虑可能存在的健康负面影响以及有益于健康的方面, 从而权衡其潜在的危险和利益。
1 转基因食品的检测技术
不论是对转基因产品进行标准管理,或是对转基因与非转 基因原料的分别输送,对食品中的转基因含量的多少加以限 制,转基因原料和食品的检测技术是必不可少的。转基因食品 的安全性检验,第一步是对受检产品进行鉴定,以区别转基因 食品与非转基因食品,筛选出在遗传分化过程中已失去转基因 特性的产品;第二步是对受检产品中导入的基因重组体构成的 变异情况进行检测,以确定其表达的忠实性及外源基因对受体 生物原基因组表达的影响。当前,国际社会对转基因食品检测 采用的技术路线主要有两条,针对外源DNA 和针对外源蛋白 质进行检测。检测要求已从定性检测提高到定量检测。 基金项目:南宁市科学技术局基金资助(20040244C) 作者简介:施向东(1968 - ) ,女,学士,主管技师,研究:食品卫生。 111 检测外源DNA 主要是以核酸为基础的PCR 检测方法, 通过PCR 技术和核酸探针的杂交检测技术,来准确快速地检 测外源基因。由于PCR 具有敏感性高的特点,且食品加工过 程中核酸变性程度不及蛋白质,因此,目前转基因食品的定量 检测仍基于PCR。主要包括半定量PCR 法、定量竞争PCR 法、实时定量PCR 法、以及PCR - EL ISA 法等。半定量PCR 和定量竞争PCR 法都是终点检测方法,前者是由标准品建立 的标准曲线来判定待测样品中转基因成分的含量。欧洲12 家
实验室验证了Huber 等[ 2 ]建立的定量竞争PCR 法,表明可检 测015 %的RRS(Rundup Ready Soybean ,抗草甘膦大豆) ,另有 实验表明以200ng DNA 为模板时,定量竞争PCR 可检出仅含 011 %转基因成分的样品[ 3 ] 。实时荧光定量PCR 技术由美国 Applied Biosystems 公司于1995 年研究出来,该技术目前成为 转基因食品定量检测中的主要检测技术。Hagen 等[ 4 ] 的研究 表明,荧光定量PCR 法可检测除脂肪、油类、调味品外,包括豆 腐等在内的豆制品中转基因成份的含量。复式及多重PCR 是 常规PCR 方法的改进,是针对多个靶位点进行同时检测,所以 其检测效果较之普通PCR 更为可靠,同时也能降低检测成本, 张平平等[ 5 ]采用该方法仍可以对含量仅为0115 %转基因大豆 进行可靠的鉴定。PCR - EL ISA 法是一种将PCR 的高效性、 高灵敏度与EL ISA 的高准确度相结合的方法,它是以地高辛 标记的特异性探针诱捕PCR 产物在合适条件下杂交,再用碱 性磷酸酯酶标记的链霉亲和素进行EL ISA 反应,既适合于快 速的定性筛选又可进行准确的定量分析。结果通过酶标仪直 接输出,无人为误差,可靠性强,自动化程度高,易于操作。方 法灵敏度高达011 % ,足以达到欧盟的要求(转基因检测阈值 为1 %) ,利用PCR - EL ISA 对转基因大豆的检测灵敏度比欧 盟推荐PCR 方法提高5,10 倍[ 6 ] 。
最近几年出现的基因芯片技术又称DNA 芯片或DNA 微 阵列技术,其能以高信息量、高通量,同时检测、分析大量的 DNA/ RNA。此项技术既可对转基因食品进行定性检测,还可 以定量的检测其种类。利用该技术可检测食用成品和鲜活的
具有灵敏性强、自动化程度高、特异性强、假阳性 动植物材料,
中国公共卫生管理2008 年第24 卷第1 期(总第127 期) Chin. J of PHM ,Feb. 2008 Vol.
24 No. 1 ?49 ?
低、简便快速的特点[ 7 ] 。基因芯片技术目前也仅是在医学、分 子生物学等少数领域得到一定应用。随着该技术的发展,成本 的降低,此技术在转基因食品的检测中极有发展前途。目前, 世界各国都在积极的进行此项研究工作,我国在“863”计划中 也拨出专项资金进行基因芯片的开发。
112 检测外源蛋白质 检测外源基因的表达产物蛋白质主要 包括酶联免疫法( EL ISA) 、蛋白质印迹法(Western blot) 、试纸 条法(Lateral Flow Strip) 和快速检测试剂盒法。 EL ISA 法用于间接检测转基因表达的目的蛋白质,该检测 法可对样品进行定性检测以及定量分析。此方法灵敏度高,易 于处理操作,但只适用于原料性食品,难于应用于加工品,因为 外源基因表达的蛋白会因加工而失活、分解或消失,增加了检 测的不确定性和较差的重复性,同时也提高了假阴性率。目 前,美国FDA 已研究出用双夹心EL ISA 法检测食品中是否含 有转基因玉米成分。
蛋白质印迹法普遍用于分离、检测特异的目的蛋白质,灵 敏度为1,5ng。Van Duijn 等人[ 8 ] 用蛋白质印迹法检测RRS
中的CP4 合成酶,检测限在015 %,1 % ,经验证这种方法可应
用于转基因大豆原材料和大豆蛋白质产品的检测。
试纸条法的原理与EL ISA 法相似,操作相对简单,且不需
要特殊实验设备,适用于现场检验或初筛。快速检测试剂盒法
也具有操作简单、使用方便的优点,但在检测结果的正确性方
面因较少的称取样量,可能会受到影响。
2 展望
随着全球经济一体化的发展,各国间贸易往来日益增加,
科技信息的频繁交流,食品安全已经跨越国界。某一地区的食
品安全问题很有可能波及全球。因而完善转基因食品的安全
监管体系,加强我国转基因食品的立法和管理工作,提高食品
安全检测技术水平迫在眉睫。目前,许多国家如欧盟、美国、日
本等国已要求中国出示非转基因产品证明,且我国每年都要从
美国、加拿大、阿根廷、澳大利亚等国进口相当数量的农产品,
这些国家正是种植转基因农作物规模较大的国家,现已经从一
些没有标识的进口食品中检测出了转基因成分。因此,为了满
足进出口国际贸易的要求,对转基因产品进行检测,已显得十
分迫切。目前,尚无适用于检测多种转基因成分的方法,还没
有国际认同的检测标准和方法,我国对转基因食品的检测工作
主要集中在检验检疫系统中,中国进出口商品检验技术研究所
建立了从食用油中提取DNA 设立转基因食品检测研究中心,
并对食用油中的转基因成分进行定性检测的方法,该方法的灵
敏度和准确性达到国际领先水平。
从国际来看,基因芯片技术和DNA 生物传感器等新技术
的研究取得了新的进展,普及利用这些新的转基因监测技术将
是今后一段时间内的研究方向之一。同时随着转基因食品种
类的日益多样化和高产量化,转基因食品的检测技术将朝着高
通量、高灵敏度、自动化和低成本化的方向发展。应该进一步
寻找快速简便精确的实验方法,加强国际合作与交流,确定通
用检测标准,让具有安全保障的转基因食品进入人民餐桌。
参考文献
[ 1 ] Millstone E ,Brunner E ,Mayer S.Beyond2substantialequivalence. Na2
ture ,1999 ,401 (6753) :525 - 526.
[2 ] Hubner P , Studer E ,Luthy J . Quatitative competitive PCR for the
detection of genetically modified organisms in food. Food Control ,
1999 :10 (6) :353 - 358.
[3 ] Wurz A ,Bluth A. Zeltz P ,et al. Quantitative analysis of genetically
modified organisms( GMO) in processed food by PCR2based meth2
ods. Food Control ,1999 ,10 (6) :385 - 389. [ 4 ] Hagen M ,Beneke B. Detection of genetically modified soy (Roundup2
Ready) in processed food products. Berl Munch Tierarztl Wochen2
schr ,2000 ,113 (6) :454 - 458.
[5 ] 张平平,刘宪华. 多重PCR 方法对大豆转基因食品的定性检测
[J ] . 食品科学,2004 ,25 (11) :227 - 230.
[ 6 ] 单宏. PCR - EL ISA 方法在转基因大豆检测中的应用. 大豆通报, 2006 ,80 (1) :37 - 39.
[ 7 ] 韩翠丽,刘代成. 基因芯片在食品检测中的应用[J ] . 生物学杂志. 2004 ,21 (1) :41.
[8 ] Gert van Duijn ,Ria van Biert ,Henrieette Bleeker2Marcelis ,etal. De2
tection methods for genetically modified crops. Food Control ,1999 ,
10 :375 - 378.
(收稿日期:2007 - 01 - 31)
【卫生监督管理】
熟肉制品生产加工现状及监督管理对策
徐政 (浙江省温岭市卫生监督所,317500)
中图分类号:R155. 62 文献标识码:A 文章编号:1001 - 9561 (2008) 01 - 0050 - 02
熟肉制品消费市场广阔,其生产加工中存在的卫生问题也
日益受到各级政府、管理部门、人大、政协关注。但从目前此行
业的现状来看,由于熟肉制品生产加工产业化程度低,缺少标
准化的生产加工企业,在城乡小作坊式生产加工点大量存在,
卫生条件差,不规范生产加工等现象给此行业的健康发展带来
较大的食品安全隐患。
1 熟肉制品生产加工形式
111 大型工业化、规模化生产加工企业 此类企业在整个行
这部分企业因在生产加工硬软件方面配备 业中只占一小部分,
都比较齐全,办理了卫生许可证。
112 超市和前店后场 在超市和前店后场中现制现卖的生产
加工模式。此种模式按照自制零售食品卫生许可条件操作,生
产加工卫生条件和生产质量有了一定的保证,生产加工场所与
生活场所严格区分,工艺流程布局合理,卫生条件、从业人员健
?50 ? 中国公共卫生管理2008 年第24 卷第1 期(总第127 期) Chin. J of PHM ,Feb.
2008 Vol. 24 No. 1
聚合酶链式反应(PCR)
原理
DNA的半保留复制时,双链DNA在多种酶的作用下可以变性解链成单链,在DNA聚合酶与启动子的参与下,根据碱基互补配对原则复制成同样的两分子挎贝。在实验条件下,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,并设计引物做启动子,加入DNA聚合酶、dNTP就可以完成特定基因的体外复制。PCR类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。
PCR由变性 - 退火(复性)- 延伸三个基本反应步骤构成:
?模板DNA的变性:模板DNA经加热至94?左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;
?模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至40~60?左右,引物与模板DNA单链的互补序列配对结合;
?引物的延伸:DNA模板 - 引物结合物在DNA聚合酶的作用下,于72?左右,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA链互
补的半保留复制链,重复循环就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2,4分钟,2,3小时就能将待扩目的基因扩增放大几百万倍。
PCR技术分类(常用)
(1)反向PCR技术(Inverse PCR, IPCR):反向PCR是克隆已知序列旁侧序列的一种方法(主要原理是用一种在已知序列中无切点的限制性内切酶消化基因组DNA(后酶切片段自身环化(以环化的DNA作为模板,用一对与已知序列两端特异性结合的引物,扩增夹在中间的未知序列。该扩增产物是线性的DNA片段,大小取决于上述限制性内切酶在已知基闲侧翼DNA序列内部的酶切位点分布情况。用不同的限制性内切酶消化,可以得到大小不同的模板DNA,再通过反向PCR获得未知片段 。
(2)锚定PCR技术(Anchored PCR, APCR):用酶法在一通用引物反转录cDNA3’-末端加上一段已知序列, 然后以此序列为引物结合位点对该cDNA进行扩增, 称为APCR。 (3)不对称PCR技术(asymmetric PCR):两种引物浓度比例相差较大的PCR技术称不对称PCR。在扩增循环中引入不同的引物浓度, 常用50,100?1比例。在最初的10,15个循环中主要产物还是双链DNA, 但当低浓度引物被消耗尽后, 高浓度引物介导的PCR反应就会产生大量单链DNA。
(4)反转录PCR技术(reverse transcription PCR, RT-PCR):当扩增模板为RNA时, 需先通过反转录酶将其反转录为cDNA才能进行扩增。应用非常广泛, 无论是分子生物学还是临床检验等都经常采用。
(5)巢式PCR技术(NEST-PCR):先用一对靶序列的外引物扩增以提高模板量, 然后再用一对内引物扩增以得到特异的PCR带, 此为巢式PCR。若用一条外引物作内引物则称之为半巢式PCR。为减少巢式PCR的操作步骤可将外引物设计得比内引物长些, 且用量较少, 同时在第一次PCR时采用较高的退火温度而第二次采用较低的退火温度, 这样在第一次PCR时, 由于较高退火温度下内引物不能与模板结合, 故只有外引物扩增产物, 经过若干次循环, 待外引物基本消耗尽, 无需取出第一次PCR产物, 只需降低退火即可直接进行PCR扩增。这不仅减少操作步骤, 同时也降低了交叉污染的机会。这种PCR称中途进退式PCR,主要用于极少量DNA模板的扩增。
(6)多重PCR技术(multiplex PCR):在同一反应中用多组引物同时扩增几种基因片段,如果基因的某一区段有缺失,则相应的电泳谱上这一区带就会消失。主要用于同一病原体的分型及同时检测多种病原体、多个点突变的分子病的诊断。
(7)重组PCR技术:重组PCR技术是在两个PCR扩增体系中, 两对引物分别由其中之一在其5’-端和3’-端引物上带上一段互补的序列,混合两种PCR扩增产物,经变性和复性,两组PCR产物互补序列发生粘连,其中一条重组杂合链能在PCR 条件下发生聚合延伸反应,产生一个包含两个不同基因的杂合基因。
(8)原位PCR技术:利用完整的细胞作为一个微小的反应体系来扩增细胞内的目的片段,在不破坏细胞的前提下,利用一些特定的检测手段来检测细胞内的扩增产物。直接用细胞涂片或石蜡包埋组织切片在单个细胞中进行,,,扩增,可进行细胞内定位,适用于检测病理切片中含量较少的靶序列。
(9)荧光定量实时PCR技术
反应动力学
PCR的三个反应步骤反复进行,使DNA扩增量呈指数上升,最终DNA扩增量可用Y=(1+X)n计算,Y代表DNA片段扩增后的拷贝数,X表示平均每次的扩增效率,n表示循环次数。平均扩增效率理论值为100%,但实际情况达不到,反应初期靶序列DNA片段的增加呈指数形式,随着PCR产物的逐渐积累,被扩增的DNA片段不再呈指数增加,而进入线性增长期或静止期,即出现“停滞效应”,此效应称平台期,与DNA聚合酶数量和活性、PCR扩增效率、非特异性产物的竞争等因素有关。
PCR扩增产物
可分为长产物片段和短产物片段两部分,短产物片段的长度严格地限定在两个引物链5’端之间,是需要扩增的特定片段。短、长产物片段是由于引物所结合的模板不同而形成的,以一个原始模板为例,在第一个反应周期中,以两条互补的DNA为模板,引物是从3’端开始延伸,其5’端是固定的,3’端则没有固定的止点,长短不一,这就是长产物片段。进入第二个反应周期后,引物除与原始模板结合外,还要同新合成的链(长产物片段)结合引物在与新链结合时,由于新链模板的5’端序列是固定的,故这次延伸的片段3’端被固定了止点,保证了新片段的起点、止点都限定于引物扩增的序列以内,形成长短一致的短产物片段。由于短产物片段是按指数倍数增加,而长产物片段则以算术倍数增加,故可忽略不计,使得PCR的反应产物不需再纯化既可以保证足够纯的DNA片段供分析、监测。
反应五要素 --- 引物、模板、DNA聚合酶、四种dNTP、Mg2+
(1)引物设计 ,
-30bp,常用为20bp左右。 ?引物长度:15
?引物扩增跨度:以200-500bp为宜,特定条件下可扩增长至10kb的片段。
?引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。ATGC最好随机分布,引物自身连续互补碱基不能超过3个,一对引物间也不易多于四个连续碱基的互补性。 【引物的GC含量和Tm值应该协调:Tm=4(G+C)+2(A+T)】
?避免引物内部出现二级结构,避免两条引物间互补,特别是3'端的互补,否则会形成引物二聚体,产生非特异的扩增条带。
?引物3’端为关键碱基,是PCR延伸的起始端,不能进行任何修饰,也不能形成二级结构的可能,一般3‘端也不能发生错配,应严格要求配对,以避免因末端碱基不配对而导致PCR失败。 5’端无严格限制,只起限定PCR产物长度的作用,对扩增特异性影响不大,因此常用来引进修饰位点或标记物。
?引物中有或能加上合适的酶切位点, 被扩增的靶序列最好有适宜的酶切位点,这对酶切分析或分子克隆很有好处。
?引物的特异性:引物应与核酸序列数据库的其它序列无明显同源性。
?引物量: 每条引物的浓度0.1,1umol或10,100pmol,以最低引物量产生所需要的结果为好,引物浓度偏高会引起错配和非特异性扩增,且可增加引物之间形成二聚体的机会。
(2)DNA聚合酶:目前有两种Taq DNA聚合酶供应,一种是从栖热水生杆菌中提纯的天然酶,另一种为大肠菌合成的基因工程酶。催化一典型的PCR反应约需酶量0.5-2.5 U/50 ml,浓度过高可引起非特异性扩增,浓度过低则合成产物量减少。
(3)dNTP的质量与浓度:dNTP溶液呈酸性,使用时应配成高浓度后,以1M NaOH或1M Tris-HCL的缓冲液将其PH调节到7.0,7.5,小量分装, -20?冰冻保存。多次冻融会使dNTP降解。在PCR反应中,dNTP应为50,200umol/L,尤其是注意4种dNTP的浓度要相等( 等摩尔配制),
如其中任何一种浓度不同于其它几种时(偏高或偏低),就会引起错配。浓度过低又会降低PCR产物的产量。
(4)模板(靶基因)核酸:模板核酸的量与纯化程度,是PCR成败与否的关键环节之一,传统的DNA纯化方法通常采用SDS和蛋白酶K来消化处理标本。SDS的主要功能是: 溶解细胞膜上的脂类与蛋白质,因而溶解膜蛋白而破坏细胞膜,并解离细胞中的核蛋白,SDS 还能与蛋白质结合而沉淀; 蛋白酶K能水解消化蛋白质,特别是与DNA结合的组蛋白,再用有机溶剂酚与氯仿抽提掉蛋白质和其它细胞组份,用乙醇或异丙醇沉淀核酸。提取的核酸即可作为模板用于PCR反应。
(5)Mg2+浓度:Mg2+对PCR扩增的特异性和产量有显著的影响,在一般的PCR反应中,各种dNTP浓度为200umol/L时,Mg2+浓度为1.5,2.0mmol/L为宜。Mg2+浓度过高,反应特异性降低,出现非特异扩增,浓度过低会降低Taq DNA聚合酶的活性,使反应产物减少。 RT-PCR -- 是将RNA的反转录(RT)和cDNA的聚合酶链式扩增(PCR)相结合的技术。 ?总RNA提取(-70?保存)
Trizol法:
1、取组织100?于玻璃匀浆器中,加入1ml Trizol 冰上抽打匀浆(边匀浆边暂停)
2、移入1.5mlEP管中,颠倒混匀,室温保存5min
3、加氯仿0.2ml(总体积的1/5),振荡混合,室温放置5min
4、4?,离心12000 rpm,15min
μl), 5、取上层液相(约400μl)移入一新1.5ml EP管中,加等体积异丙醇(约400振荡混合,室温保存10min
6、4?,离心12000 rpm,10min
7、弃上清液,加1ml 75%无水乙醇(4?保存),振荡混合
8、4?,离心7500 rpm,5min
9、弃上清液,室温放置20min(EP管倒置在滤纸上)使RNA沉淀干燥(不能完全干燥)
10、加20ul DEPC水至EP管中,在60?孵育3-5min(或手握3-5min),使RNA充分溶解( 可保存在液氮或低温冰箱-70?)
测RNA浓度:走RNA变性电泳观察18s、28s条带,分光光度计检测260/280吸光度比值
调零:750ul DEPC水
测量:745ul DEPC水 + 5ul RNA
总RNA浓度= OD260×40×稀释倍数(150倍)ug/ml
= OD260×40×150 ug/ml
= OD260×6 ug/ul
A1/A2应在1.8-2.0之间
注意:提取RNA的质量主要是要通过260/280的比值看是不是在2.0左右,当OD260/OD280<1.8时提示有蛋白或酚的污染,需要进一步纯化rna;还要跑胶看看28s、18s、5s的三条带是不是清晰,一般质量好的rna,28s:18s的亮度比例在2:1左右,最后一条5s的应该比较淡。>
?逆转录RT(cDNA:一周内-20?保存,长期-70?保存)
RT反应体系:20ul体系
第一步:约20分钟
RNA抽提物 5μl
Radome引物 2μl
RNAsin 0.5μl
1、65? 15分钟
2、立即放入冰浴
第二步:约2小时
RNAsin 0.5μl
10mM dNTP 1μl
5× RT缓冲液 4μl
25mM MgCl2 4μl
AMV 3μl
1、37? 1.5小时
2、94? 5-10分钟
3、反应物保存于-20?或进行PCR
?聚合酶链反应PCR(产物-20?保存)
PCR反应体系:100μl体系
约4.5小时 约5小时
25mM MgCl2 2μl 3μl
10×PCR缓冲液 5μl 5μl
10mM dNTP 1μl 1μl
10pmol/μl上游引物 2.5μl 2.5μl
10pmol/μl下游引物 2.5μl 2.5μl
cDNA模板 2.5μl 5μl
ddH2O 34μl 30μl
Taq酶 0.5μl 1μl
轻质石蜡油 50μl 50μl
1、94? 2分钟,55? 1分钟,72? 2分钟 为第一个步1个循环
2、94? 45秒,55? 40秒,72? 1分钟 为第二步30/40个循环
3、72? 10分钟
4、取出后4?保存,5分钟后-20?保存或走电泳
?琼脂糖凝胶电泳:约1.25小时
1、配制0.5× TBE电泳缓冲液300 ml
2、配制凝胶浓度1.7%(40ml 0.5×TBE加0.68g胶),微波炉中火2分钟溶解胶,冷却
至60?加入溴乙锭2μl(10mg/ml的终浓度0.5μg/ml),充分混匀
3、将温热的凝胶倒入已置好梳子的胶膜中,在室温下放置30-45min后现进行电泳
4、加样8μl PCR反应产物+2μl溴酚兰 ,空一格加DNA marker
5、电泳50-80V ,电场强度不宜高于5V/cm
6、在紫外灯下观察,DNA 存在则显示出红色荧光条带,采用凝胶成像系 统拍照保存。
电泳缓冲液(5×TBE贮存液):Tris 54g、硼酸27.5g、0.5M EDTA 20ml pH8.0、加蒸溜水至1000ml。使用时,将5×TBE稀释10倍成0.5×TBE就可以在电泳时使用(工作浓度)。
上样缓冲液(6×缓冲液,4?保存):0.25%溴酚蓝、0.25%二甲苯青FF、30%甘油
琼脂糖凝胶配制:(1.0%)1.0g琼脂糖+100ml电泳缓冲液,微波炉加热至沸腾(如果首次煮胶,一定要煮透,以不出现泡沫为标志),熔化的琼脂物冷却至60?时加入10mg/ml溴化乙锭5μl,充分混匀,将温热的凝胶倒入已置好梳子的胶膜中,在室温下放置30-45min(可以放入4度冰箱加快琼脂糖凝固)后现进行电泳。1.5%同上,将琼脂糖的量改为1.5g。
试剂:
DEPC水,1、DEPC水:吸出1ml DEPC放在1000ml容量瓶中加双蒸水定容至1000ml,配成1‰并充分振荡混匀备用。
2、75%乙醇:用无水乙醇+DEPC水配,然后放-20?保存。
(DEPC水需先高压,高压时注意:1.装DEPC水的瓶子在盖子和瓶之间要放上线头;2.高压时间在40-45分钟,所以水要适当多加一点;3.高压结束后,不要强行放气,要让压力自然下降,不然水会喷出)。
3、异丙醇:放入棕色瓶中。
4、氯仿:放入棕色瓶中。
5、轻质石蜡油
Trizol:一种新型总RNA抽提试剂,可以直接从细胞或组织中提取总RNA。其含有苯酚、异硫氰酸胍等物质,能迅速破碎细胞并抑制细胞释放出的核酸酶。
DEPC:焦碳酸二乙酯,是一种强烈但不彻底的RNA酶抑制剂
随机引物、Oligo(dT)、基因特异性引物:用于反转录的引物可视实验的具体情况选择
M-MLV、AMV(配10×RT-buffer):逆转录酶
RNasin:RNA酶抑制剂
dNTP:脱氧核糖核苷三磷酸(dATP、dGTP、dTTP、dCTP、dUTP)
Taq酶(配10×PCR-buffer、MgCl2):DNA聚合酶
Primer(上游、下游)
Marker:是分子量不同的DNA片段,主要用途就是DNA 分子凝胶电泳时,加样用做对比来检测琼脂糖凝胶是否有问题。
附:TBE(Tris硼酸缓冲液)
范文五:pcr反应体系
标准的 PCR 反应体系:
10×扩增缓冲液 10ul
4种 dNTP 混合物 各 200umol/L
引物 各 10~100pmol
模板 DNA 0.1~2ug
Taq DNA 聚合酶 2.5u
Mg2+1.5mmol/L
加双或三蒸水至 100ul
PCR 反应五要素
参加 PCR 反应的物质主要有五种即引物、酶、 dNTP 、模板和 Mg2+
引物 :引物是 PCR 特异性反应的关键, PCR 产物的特异性取决于引物与模板 DNA 互补的程度。理论上,只要知道任何一段模板 DNA 序列, 就能 按其设计互补的寡核苷酸链做引物,利用 PCR 就可将模板 DNA 在体外大量扩增。
设计引物应遵循以下原则:
①引物长度:15-30bp ,常用为 20bp 左右。
②引物扩增跨度:以 200-500bp 为宜,特定条件下可扩增长至 10kb 的片段。
③引物碱基:G+C含量以 40-60%为宜, G+C太少扩增效果不佳, G+C过多易出现非特异条带。 ATGC 最好随机分布,避免 5个以上的嘌呤或嘧啶 核苷酸 的成串排列。
④避免引物内部出现二级结构,避免两条引物间互补,特别是 3' 端的互补,否则会形成引物二聚体,产生非特异的扩增条带。
⑤引物 3' 端的碱基,特别是最末及倒数第二个碱基,应严格要求配对,以避免因末端碱基不配对而导致 PCR 失败。
⑥引物中有或能加上合适的酶切位点, 被扩增的靶序列最好有适宜的酶切位点, 这对酶切分析或分子克隆很有好处。
⑦引物的特异性:引物应与核酸序列数据库的其它序列无明显同源性。引物量:每条引物的浓度 0.1~1umol 或 10~100pmol ,以最低引物 量产生所需要的 结果为好,引物浓度偏高会引起错配和非特异性扩增,且可增加引物之间形成二聚体的机会。
酶及其浓度 目前有两种 Taq DNA 聚合酶供应, 一种是从栖热水生杆菌中提纯的天然酶,另一种为大肠菌合成的基因工程酶。催化一典型的 PCR 反应约需酶量 2。 5U(指总反应体积为 100ul 时 ) ,浓度过高可引起非特异性扩增,浓度过低则合成产物量减少。
dNTP 的质量与浓度 dNTP 的质量与浓度和 PCR 扩增效率有密切关系, dNTP 粉呈颗粒状,如保存不当易变性失去生物学活性。 dNTP 溶液呈酸性, 使用时应配成高浓度后,以 1M NaOH 或 1M Tris 。 HCL 的缓冲液将其 PH 调节到 7.0~7.5,小量分装, -20℃冰冻保存。多次冻融会使 dNTP 降解。在 PCR 反应中, dNTP 应为 50~200umol/L,尤其是注意 4种 dNTP 的浓度要相等 ( 等摩尔配制 ) ,如其中任何一种浓度不同于其它几种时 (偏高或偏低 ) ,就会引起错 配。浓度过低又会降低 PCR 产物的产量。 dNTP 能与 Mg2+结合,使游离的 Mg2+浓度降低。
模板 (靶基因 ) 核酸 模板核酸的量与纯化程度,是 PCR 成败与否的关键环节之一,传统的 DNA 纯化方法通常采用 SDS 和蛋白酶 K 来消化处理标本。 SDS 的主要功能是:溶解细胞膜上的脂类与蛋白质,因而溶解膜蛋白而破坏细胞膜,并解离细胞中的核蛋白, SDS 还能与蛋白质结合而沉淀; 蛋白酶 K 能 水解消化蛋白质,特别是与 DNA 结合的组蛋白,再用有机溶剂酚与氯仿抽提掉蛋白质和其它细胞组份,用乙醇或异丙醇沉淀核酸。提取的核酸即可作为模板用 于 PCR 反应。一般临床检测标本,可采用快速简便的方法溶解细胞,裂解病原体,消化除去染色体的蛋白质使靶基因游离,直接用于 PCR 扩增。 RNA 模板提 取一般采用异硫氰酸胍或蛋白酶 K 法,要防止 RNase 降解 RNA 。
Mg2+浓度 Mg2+对 PCR 扩增的特异性和产量有显著的影响,在一般的 PCR 反应中,各种 dNTP 浓度为 200umol/L时, Mg2+浓度为 1.5~2.0mmo l/L为宜。 Mg2+浓度过高,反应特异性降低,出现非特异扩增,浓度过低会降低 Taq DNA 聚合酶的活性,使反应产物减少。
PCR 反应条件的选择
PCR 反应条件为温度、时间和循环次数。
温度与时间的设置:基于 PCR 原理三步骤而设置变性 -退火 -延伸三个温度点。在标准反应中采用三温度点法,双链 DNA 在 90~95℃变性,再 迅速冷却至 40 ~60℃,引物退火并结合到靶序列上,然后快速升温至 70~75℃,在 Taq DNA 聚合酶的作用下,使引物链沿模板延伸。对于较短靶基因 (长 度为 100~300bp 时 ) 可采用二温度点法, 除变性温度外、退火与延伸温度可合二为一,一般采用 94℃变性, 65℃左右退火与延伸 (此温度 Taq DNA 酶仍有较 高的催化活性 ) 。
①变性温度与时间:变性温度低, 解链不完全是导致 PCR 失败的最主要原因。 一般情况下, 93℃~94℃ lmin 足以使模板 DNA 变性, 若低于 93℃ 则需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或 PCR 产物完全变性,就会导致 PCR 失败。
②退火 (复性 ) 温度与时间:退火温度是影响 PCR 特异性的较重要因素。变性后温度快速冷却至 40℃~60℃,可使引物和模板发生结合。由于模 板 DNA 比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱基组成及其浓度,还有 靶基序列的长度。对于 20个核苷酸, G+C含量约 50%的引物, 55℃为选择最适退火温度的起点较为理想。引物的复性温度可通过以下公式帮助选择合适的温 度:
Tm 值 (解链温度 )=4(G+C)+2(A+T)
复性温度 =Tm值 -(5~10℃ )
在 Tm 值允许范围内, 选择较高的复性温度可大大减少引物和模板间的非特异性结合, 提高 PCR 反应的特异性。复性时间一般为 30~60sec , 足以使引物与模板之间完全结合。
③延伸温度与时间:Taq DNA 聚合酶的生物学活性:
70~80℃ 150核苷酸 /S/酶分子
70℃ 60核苷酸 /S/酶分子
55℃ 24核苷酸 /S/酶分子
高于 90℃时, DNA 合成几乎不能进行。
PCR 反应的延伸温度一般选择在 70~75℃之间,常用温度为 72℃,过高的延伸温度不利于引物和模板的结合。 PCR 延伸反应的时间,可根据待 扩增片段的长度而定,一般 1Kb 以内的 DNA 片段,延伸时间 1min 是足够 的。 3~4kb 的靶序列需 3~4min ;扩增 10Kb 需延伸至 15min 。延伸进间过长会导 致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。
循环次数 循环次数决定 PCR 扩增程度。 PCR 循环次数主要取决于模板 DNA 的浓度。一般的循环次数选在 30~40次之间,循环次数越多,非特 异性产物的量亦随之增多。
转载请注明出处范文大全网 » pcr反应步骤及注意事项

